



1. ความเข้าใจเบื้องต้น
ในขั้นตอนนี้ เราต้องเข้าใจแนวคิดและคำศัพท์บางคำ เพื่อไม่ให้ทำผิดต่อหน้ารุ่นพี่ เช่น
ถาม: อะไรคือความแตกต่างระหว่าง RT-PCR, qPCR, Real-time PCR และ real-time RT-PCR?
คำตอบ: RT-PCR คือ PCR การถอดความแบบย้อนกลับ(PCR การถอดความแบบย้อนกลับ, RT-PCR) ซึ่งเป็นตัวแปรของปฏิกิริยาลูกโซ่โพลิเมอเรส (PCR) ที่ใช้กันอย่างแพร่หลายใน RT-PCR สาย RNA จะถูกแปลงย้อนกลับเป็น DNA เสริม ซึ่งจากนั้นจะใช้เป็นแม่แบบสำหรับการขยาย DNA โดย PCR
PCR แบบเรียลไทม์และ qPCR(Quantitative Rea-ltime-PCR) เป็นสิ่งเดียวกัน ทั้งคู่เป็น PCR เชิงปริมาณตามเวลาจริง ซึ่งหมายความว่าแต่ละรอบของ PCR มีการบันทึกข้อมูลแบบเรียลไทม์ ดังนั้นจำนวนเทมเพลตเริ่มต้นจึงสามารถปรับการวิเคราะห์ได้อย่างแม่นยำ
แม้ว่าทั้ง Real-time PCR (real-time fluorescent quantitative PCR) และ Reverse transcription PCR (reverse transcription PCR) ดูเหมือนจะเรียกโดยย่อว่า RT-PCR แต่อนุสัญญาระหว่างประเทศคือ: RT-PCR หมายถึงการถอดความแบบย้อนกลับโดยเฉพาะPCR , Real-time PCR โดยทั่วไปย่อมาจาก qPCR (PCR แบบเรียลไทม์เชิงปริมาณ).
และเรียลไทม์ RT-PCR (RT-qPCR) เป็น PCR ถอดรหัสย้อนกลับรวมกับเทคโนโลยีเชิงปริมาณเรืองแสง: ก่อนอื่นขอรับ cDNA (RT) จากการถอดรหัสย้อนกลับของ RNA จากนั้นใช้ PCR แบบเรียลไทม์สำหรับการวิเคราะห์เชิงปริมาณ (qPCR)ห้องปฏิบัติการส่วนใหญ่ทำ RT-qPCR นั่นคือการวิจัยเกี่ยวกับการควบคุมการแสดงออกของ RNA ดังนั้น qPCR ที่ทุกคนพูดถึงในห้องปฏิบัติการจึงหมายถึง RT-qPCR แต่อย่าลืมว่ายังมีการทดสอบ DNA จำนวนมากในการใช้งานทางคลินิกการวิเคราะห์เชิงปริมาณ เช่น การตรวจหาไวรัสตับอักเสบบี ไวรัสตับอักเสบบี
คำถาม: หลังจากอ่าน PCR เชิงปริมาณฟลูออเรสเซนต์จำนวนมาก เหตุใดจึงควรควบคุมส่วนขยายให้อยู่ในช่วง 80-300bp
คำตอบ: ความยาวของลำดับยีนแต่ละลำดับจะแตกต่างกัน บางตัวยาวหลาย kb บางตัวยาวเป็นร้อย bp แต่เราต้องการเพียงความยาวผลิตภัณฑ์ 80-300bp เมื่อออกแบบไพรเมอร์ สั้นหรือยาวเกินไปไม่เหมาะสำหรับการตรวจจับ PCR เชิงปริมาณเรืองแสงชิ้นส่วนของผลิตภัณฑ์สั้นเกินไปที่จะแยกความแตกต่างจากไพรเมอร์-ไดเมอร์ความยาวของไพรเมอร์-ไดเมอร์อยู่ที่ประมาณ 30-40bp และเป็นการยากที่จะแยกแยะว่าเป็นไพรเมอร์-ไดเมอร์หรือผลิตภัณฑ์หากมีค่าน้อยกว่า 80bpหากชิ้นส่วนของผลิตภัณฑ์ยาวเกินไป เกิน 300bp จะทำให้ประสิทธิภาพการขยายต่ำและไม่สามารถตรวจจับปริมาณของยีนได้อย่างมีประสิทธิภาพ
ตัวอย่างเช่น เมื่อคุณนับจำนวนคนในห้องเรียน คุณต้องนับจำนวนปากเท่านั้นเช่นเดียวกับเมื่อคุณตรวจหายีน คุณจะต้องตรวจหาลำดับของยีนบางลำดับเท่านั้นจึงจะแสดงถึงลำดับทั้งหมดได้ถ้าจะนับคน ต้องนับทั้งปาก จมูก หู และแว่นตา ผิดพลาดได้ง่าย
เพื่อขยายขอบเขต ในการวิจัยทางชีววิทยา มีกรณีการวิจัยมากมายจากจุดหนึ่งไปยังอีกพื้นที่หนึ่ง เนื่องจากลำดับยีนของสปีชีส์ใดๆ นั้นยาวมาก จึงไม่จำเป็นและเป็นไปไม่ได้ที่จะวัดชิ้นส่วนทั้งหมด เช่น การหาลำดับ 16S ของแบคทีเรีย ซึ่งเป็นการดำเนินการในลำดับอนุรักษ์นิยมของแบคทีเรีย Assays เพื่ออนุมานจำนวนประชากรของแบคทีเรียจำนวนหนึ่ง
ถาม: ความยาวที่เหมาะสมที่สุดสำหรับการออกแบบไพรเมอร์ qPCR คือเท่าใด
คำตอบ: โดยทั่วไป ความยาวของไพรเมอร์จะอยู่ที่ประมาณ 20-24bp ซึ่งจะดีกว่าแน่นอน เราต้องใส่ใจกับค่า TM ของไพรเมอร์เมื่อออกแบบไพรเมอร์ เนื่องจากสิ่งนี้เกี่ยวข้องกับอุณหภูมิการหลอมที่เหมาะสมที่สุดหลังจากการทดลองหลายครั้ง ได้รับการพิสูจน์แล้วว่า 60°C เป็นค่า TM ที่ดีกว่าหากอุณหภูมิการหลอมต่ำเกินไป จะนำไปสู่การขยายที่ไม่เฉพาะเจาะจงได้ง่ายหากอุณหภูมิการหลอมสูงเกินไป ประสิทธิภาพการขยายจะค่อนข้างต่ำ จุดสูงสุดของเส้นโค้งการขยายจะเริ่มขึ้นในภายหลัง และค่า CT จะล่าช้า
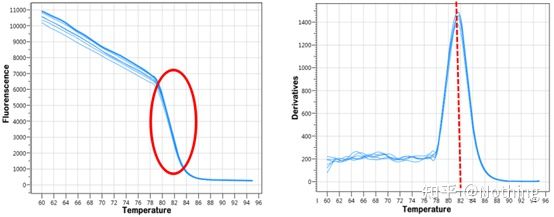
ถาม: วิธีย้อมแตกต่างจากวิธีโพรบอย่างไร?
ตอบ วิธีการย้อมสีย้อมเรืองแสงบางชนิด เช่น SYBR Green Ⅰ, PicoGreen, BEBO ฯลฯ ไม่เปล่งแสงด้วยตัวเอง แต่จะเปล่งแสงหลังจากจับกับร่องเล็ก ๆ ของ DNA ที่มีเกลียวคู่ดังนั้นในช่วงเริ่มต้นของปฏิกิริยา PCR เครื่องจึงไม่สามารถตรวจจับสัญญาณเรืองแสงได้เมื่อปฏิกิริยาไปถึงขั้นตอนการหลอม-ขยาย สายเกลียวคู่จะถูกเปิดออก และสายใหม่จะถูกสังเคราะห์ภายใต้การทำงานของ DNA พอลิเมอเรส และโมเลกุลเรืองแสงจะจับกับร่องรองของ dsDNAเมื่อจำนวนของรอบ PCR เพิ่มขึ้น สีย้อมจำนวนมากขึ้นจะถูกรวมเข้ากับ DNA แบบสองเส้น และสัญญาณเรืองแสงก็เพิ่มขึ้นอย่างต่อเนื่องเช่นกันวิธีการย้อมส่วนใหญ่จะใช้ในการวิจัยทางวิทยาศาสตร์
PS: ระวังเมื่อทำการทดลอง สีย้อมจะต้องรวมกับ DNA ของมนุษย์ ระวังจะทำให้มันกลายเป็นคนเรืองแสง
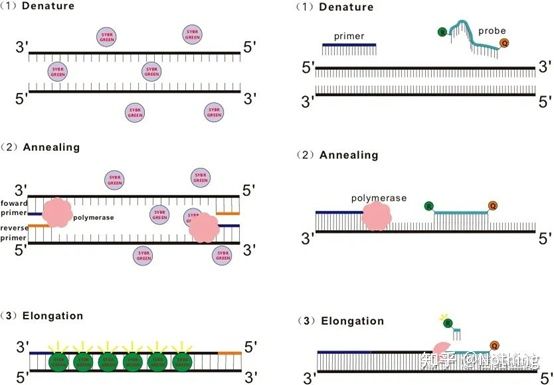
วิธีย้อมสี (ซ้าย) วิธีโพรบ (ขวา)
PS: ระวังเมื่อทำการทดลอง สีย้อมจะต้องรวมกับ DNA ของมนุษย์ ระวังจะทำให้มันกลายเป็นคนเรืองแสง
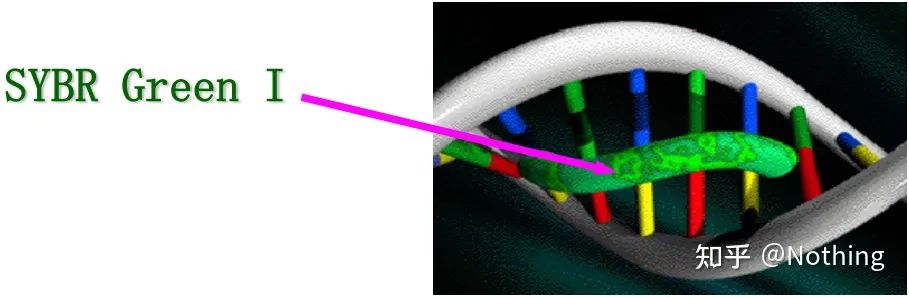
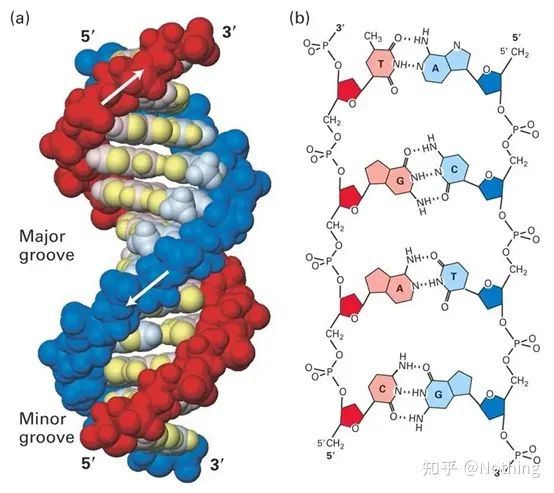
SYBR Green Ⅰ จับกับร่องเล็ก ๆ ของ DNA
วิธีการสอบสวนโพรบ Taqman เป็นโพรบไฮโดรไลซิสที่ใช้บ่อยที่สุดมีกลุ่มเรืองแสงที่ปลายโพรบ 5 ′โดยปกติจะเป็น FAM และโพรบเองก็เป็นลำดับที่เสริมกับยีนเป้าหมายมีกลุ่มดับเรืองแสงที่ปลาย 3 'ตามหลักการของการถ่ายโอนพลังงานเรโซแนนซ์ฟลูออเรสเซนซ์ (Förster resonance energy transfer, FRET) เมื่อกลุ่มนักข่าวเรืองแสง (โมเลกุลเรืองแสงของผู้บริจาค) และกลุ่มเรืองแสงดับ (โมเลกุลเรืองแสงตัวรับ) ตื่นเต้น เมื่อสเปกตรัมเหลื่อมกันและระยะทางใกล้มาก (7-10 นาโนเมตร) การกระตุ้นของโมเลกุลผู้บริจาคสามารถกระตุ้นให้เกิดการเรืองแสงของโมเลกุลตัวรับ ในขณะที่การเรืองแสงอัตโนมัตินั้น อ่อนแอลงดังนั้นที่จุดเริ่มต้นของปฏิกิริยา PCR เมื่อโพรบว่างและไม่เสียหายในระบบ กลุ่มสารเรืองแสงนักข่าวจะไม่ปล่อยสารเรืองแสงเมื่อทำการหลอม ไพรเมอร์และโพรบจะเชื่อมกับแม่แบบในระหว่างขั้นตอนการขยาย พอลิเมอเรสจะสังเคราะห์สายโซ่ใหม่อย่างต่อเนื่องDNA polymerase มีกิจกรรม exonuclease 5′-3′เมื่อไปถึงโพรบ DNA พอลิเมอเรสจะไฮโดรไลซ์โพรบจากแม่แบบ แยกกลุ่มเรืองแสงรีพอร์ตออกจากกลุ่มเควนเชอร์ฟลูออเรสเซนต์ แล้วปล่อยสัญญาณเรืองแสงเนื่องจากโพรบและแม่แบบมีความสัมพันธ์แบบหนึ่งต่อหนึ่ง วิธีการโพรบจึงดีกว่าวิธีย้อมในแง่ของความแม่นยำและความไวของการทดสอบส่วนใหญ่ใช้วิธีโพรบในการวินิจฉัย
ถาม: การหาปริมาณสัมบูรณ์คืออะไรปริมาณสัมพัทธ์คืออะไร?
คำตอบ: การหาปริมาณแบบสัมบูรณ์หมายถึงการคำนวณจำนวนสำเนาเริ่มต้นของตัวอย่างที่จะทดสอบโดย qPCR เช่น จำนวนไวรัส HBV ในเลือด 1 มล.ผลลัพธ์ที่ได้จากการหาปริมาณสัมพัทธ์คือการเปลี่ยนแปลงในปริมาณของยีนเป้าหมายในตัวอย่างเฉพาะที่สัมพันธ์กับตัวอย่างอ้างอิงอื่น และการแสดงออกของยีนจะถูกควบคุมขึ้นหรือควบคุมลง
ถาม: ปริมาณของการสกัด RNA ประสิทธิภาพการถอดรหัสย้อนกลับ และประสิทธิภาพการขยายจะส่งผลต่อผลการทดลองหรือไม่
ถาม: การเก็บตัวอย่าง รีเอเจนต์ในการสกัด รีเอเจนต์การถอดรหัสแบบย้อนกลับ และวัสดุสิ้นเปลืองที่ส่งผ่านแสงจะส่งผลต่อผลการทดลองหรือไม่
ถาม: วิธีใดที่สามารถแก้ไขข้อมูลการทดลองได้
เกี่ยวกับปัญหาเหล่านี้ เราจะอธิบายโดยละเอียดในส่วนขั้นสูงและขั้นสูงด้านล่าง
2. ความรู้ขั้นสูง
สำหรับ PCR เชิงปริมาณฟลูออเรสเซนต์ตามเวลาจริง เราต้องตระหนักถึงความเป็นจริงที่งานวิจัยทางวิทยาศาสตร์หลายพันฉบับได้รับการตีพิมพ์ทุกปี ซึ่งเทคโนโลยี PCR เชิงปริมาณฟลูออเรสเซนต์ก็ไม่ใช่จำนวนน้อยๆ
หากไม่มีมาตรฐานทั่วไปในการวัดการทดลอง PCR เชิงปริมาณด้วยฟลูออเรสเซนต์ ผลลัพธ์อาจแตกต่างกันอย่างมากสำหรับยีนเดียวกันในสปีชีส์เดียวกัน ด้วยวิธีการประมวลผลแบบเดียวกัน ผลการตรวจจับก็จะแตกต่างกันอย่างมาก และมันจะยากสำหรับผู้มาทีหลังที่จะทำซ้ำผลลัพธ์เดิมคุณไม่มีใครรู้ว่าสิ่งใดถูกและสิ่งใดผิด
นี่หมายความว่า PCR เชิงปริมาณเรืองแสงเป็นเทคโนโลยีโกงหรือไม่น่าเชื่อถือ?ไม่ เป็นเพราะ PCR เชิงปริมาณของฟลูออเรสเซนต์มีความไวและแม่นยำกว่า และการทำงานที่ผิดพลาดเพียงเล็กน้อยจะให้ผลลัพธ์ที่ตรงกันข้ามอย่างสิ้นเชิงการสูญเสียเล็กน้อยอยู่ห่างออกไปหนึ่งพันไมล์.ผู้เขียนบทความอาจถูกผู้วิจารณ์ทรมานซ้ำแล้วซ้ำเล่าในขณะเดียวกัน ผู้ตรวจสอบวารสารก็ยากที่จะเลือกจากผลการทดลองที่แตกต่างกัน
สรุปแล้วชี้ให้เห็นถึงการขาดฉันทามติในการทดลอง PCR แบบเรียลไทม์ด้วยเหตุนี้ นักวิทยาศาสตร์อาวุโสในอุตสาหกรรมจึงเริ่มกำหนดมาตรฐานกำหนดให้ผู้ร่วมให้ข้อมูลระบุรายละเอียดการทดลองและการประมวลผลข้อมูลที่จำเป็น (รวมถึงข้อมูลที่จำเป็น) ในบทความเพื่อให้เป็นไปตามมาตรฐานเหล่านี้
ผู้ตรวจสอบสามารถตัดสินคุณภาพของการทดสอบได้โดยอ่านรายละเอียดเหล่านี้ผู้อ่านในอนาคตสามารถใช้สิ่งนี้เพื่อทำซ้ำการทดสอบหรือปรับปรุงการทดสอบจากนั้นผลการทดลองที่ได้ในลักษณะนี้จะมีข้อมูลครบถ้วน มีคุณภาพสูง และใช้งานได้จริง
MIBBI (ข้อมูลขั้นต่ำสำหรับการตรวจสอบทางชีวภาพและชีวการแพทย์ -http://www.mibbi.org) เกิดขึ้นมาMIBBI เป็นโครงการที่ให้มาตรฐานสำหรับการทดลอง.มีการเผยแพร่ในธรรมชาติโครงการนี้มุ่งเป้าไปที่การทดลองทางชีววิทยาต่างๆ รวมถึงชีววิทยาของเซลล์, Microarray, qPCR ที่เราจะพูดถึงตอนนี้ ฯลฯ และจัดเตรียมการทดลองแต่ละประเภทเมื่อส่งต้นฉบับควรให้ข้อมูลนั้นตลอดเวลา
ในโครงการ MIBBI มีสองบทความที่เกี่ยวข้องกับ PCR เชิงปริมาณเรืองแสง ได้แก่:
·RDML (Real-Time PCR Data Markup Language) – ภาษาที่มีโครงสร้างและคู่มือการรายงานสำหรับข้อมูล PCR เชิงปริมาณแบบเรียลไทม์
·MIQE (ข้อมูลขั้นต่ำสำหรับการเผยแพร่การทดลอง PCR แบบเรียลไทม์เชิงปริมาณ) – ข้อมูลขั้นต่ำสำหรับการเผยแพร่บทความเกี่ยวกับการทดลอง PCR เชิงปริมาณแบบเรียลไทม์
ขั้นแรก เรามาพูดถึง RDML ซึ่งเป็นข้อกำหนดเฉพาะของคำศัพท์
หากไม่มีคำจำกัดความมาตรฐานสำหรับทุกสิ่ง การอภิปรายจะดำเนินต่อไปไม่ได้ ซึ่งเป็นเหตุผลว่าทำไมคำอธิบายคำศัพท์จึงสำคัญมากในการสอบ
คำศัพท์ที่ใช้ในการทดลอง PCR เชิงปริมาณเรืองแสงประกอบด้วยเนื้อหาต่อไปนี้QIAGEN ได้ทำการสรุปที่ดีที่สุดสำหรับเราต่อไปนี้จะแห้งทั้งหมดสินค้า .
เส้นโค้งการขยาย
เส้นโค้งการขยายหมายถึงเส้นโค้งที่เกิดขึ้นระหว่างกระบวนการ PCR โดยมีหมายเลขรอบเป็น abscissa และความเข้มของฟลูออเรสเซนตามเวลาจริงระหว่างปฏิกิริยาเป็นลำดับ
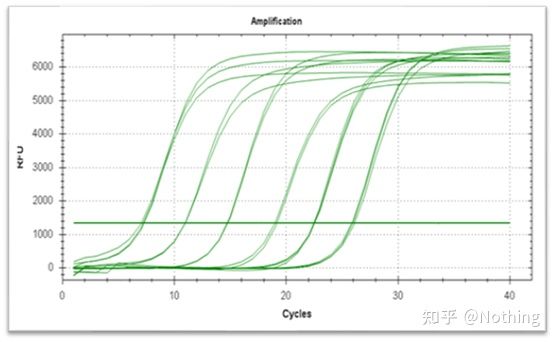
เส้นโค้งการขยายสัญญาณที่ดีเยี่ยมควรมีลักษณะดังต่อไปนี้: เส้นฐานทรงตัวหรือลดลงเล็กน้อย และไม่มีแนวโน้มขาขึ้นที่ชัดเจนจุดเปลี่ยนของเส้นโค้งมีความชัดเจน และความชันของเฟสเอกซ์โปเนนเชียลเป็นสัดส่วนกับประสิทธิภาพการขยายสัญญาณยิ่งมีความชันมากเท่าใด ประสิทธิภาพการขยายก็จะยิ่งสูงขึ้นเท่านั้นเส้นโค้งการขยายโดยรวม ความขนานเป็นสิ่งที่ดีซึ่งบ่งชี้ว่าประสิทธิภาพการขยายของแต่ละหลอดมีความคล้ายคลึงกันเฟสเอ็กซ์โปเนนเชียลของเส้นโค้งการขยายของตัวอย่างที่มีความเข้มข้นต่ำนั้นชัดเจน
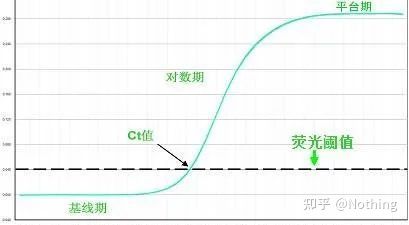
บรรทัดฐาน (บรรทัดฐาน)
ค่าพื้นฐานคือระดับเสียงของรอบต้นโดยปกติจะวัดระหว่างรอบที่ 3 และ 15 เนื่องจากไม่สามารถตรวจจับการเพิ่มขึ้นของค่าการเรืองแสงที่เกิดจากผลิตภัณฑ์ขยายสัญญาณได้ในช่วงเวลานี้จำนวนรอบที่ใช้ในการคำนวณพื้นฐานสามารถเปลี่ยนแปลงได้และอาจจำเป็นต้องลดลงหากใช้จำนวนเทมเพลตสูงหรือหากระดับการแสดงออกของยีนเป้าหมายสูง
การตั้งค่าเส้นฐานจำเป็นต้องดูข้อมูลฟลูออเรสเซนซ์จากเส้นโค้งการขยายความเป็นเชิงเส้นค่าพื้นฐานถูกตั้งค่าเพื่อให้การเติบโตของเส้นกราฟการขยายเริ่มต้นด้วยหมายเลขไซเคิลที่มากกว่าหมายเลขไซเคิลบนของเบสไลน์จำเป็นต้องตั้งค่าพื้นฐานแยกกันสำหรับแต่ละลำดับเป้าหมายค่าฟลูออเรสเซนต์เฉลี่ยที่ตรวจพบในรอบแรกจำเป็นต้องหักออกจากค่าฟลูออเรสเซนซ์ที่ได้จากผลิตภัณฑ์ขยายซอฟต์แวร์ Real-Time PCR เวอร์ชันล่าสุดช่วยให้สามารถปรับแต่งการตั้งค่าพื้นฐานสำหรับแต่ละตัวอย่างได้โดยอัตโนมัติ
ในช่วงสองสามรอบแรกของปฏิกิริยาการขยายสัญญาณ PCR สัญญาณการเรืองแสงจะไม่เปลี่ยนแปลงมากนักการเข้าใกล้เส้นตรงเรียกว่าเส้นฐาน แต่ถ้าเราดูอย่างใกล้ชิดที่สองสามรอบแรก เราจะเห็นว่าภายในเส้นฐานคือสิ่งที่เกิดขึ้นในภาพด้านล่าง
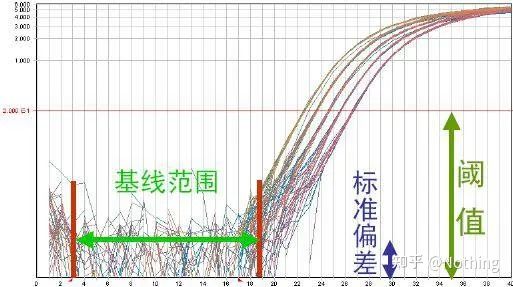
พื้นหลัง พื้นหลังหมายถึง
ค่าการเรืองแสงที่ไม่จำเพาะในปฏิกิริยาตัวอย่างเช่น: การดับเรืองแสงที่ไม่มีประสิทธิภาพหรือแม่แบบดีเอ็นเอเกลียวคู่จำนวนมากเนื่องจากการใช้ SYBR Greenส่วนประกอบพื้นหลังของสัญญาณจะถูกลบออกทางคณิตศาสตร์โดยอัลกอริทึมซอฟต์แวร์ Real-Time PCR
สัญญาณนักข่าว
สัญญาณนักข่าวหมายถึงสัญญาณเรืองแสงที่สร้างโดย SYBR Green หรือโพรบเฉพาะลำดับที่มีป้ายกำกับเรืองแสงในระหว่าง PCR แบบเรียลไทม์
สัญญาณนักข่าวที่ปรับมาตรฐาน (RN)
RN หมายถึงความเข้มของการเรืองแสงของสีนักข่าวหารด้วยความเข้มของการเรืองแสงของสีย้อมอ้างอิงแบบพาสซีฟที่วัดในแต่ละรอบ
สีย้อมอ้างอิงแบบพาสซีฟ
ใน PCR แบบเรียลไทม์บางรายการROX สีย้อมเรืองแสงใช้เป็นข้อมูลอ้างอิงภายในเพื่อทำให้สัญญาณเรืองแสงเป็นปกติ.โดยจะแก้ไขความผันแปรเนื่องจากการปิเปตที่ไม่ถูกต้อง ตำแหน่งหลุม และความผันผวนของฟลูออเรสเซนซ์ในแต่ละหลุม

เกณฑ์การเรืองแสง (เกณฑ์)
ถูกปรับให้อยู่เหนือค่าพื้นหลังและต่ำกว่าค่าที่ราบสูงของเส้นโค้งการขยายอย่างมากต้องอยู่ในขอบเขตเชิงเส้นของเส้นโค้งการขยาย ซึ่งแสดงถึงช่วงบันทึกเชิงเส้นของการตรวจจับ PCRเกณฑ์ควรตั้งค่าในมุมมองเส้นโค้งการขยายบันทึกเพื่อให้สามารถระบุเฟสบันทึกเชิงเส้นของ PCR ได้ง่ายหากมียีนเป้าหมายหลายตัวใน Real-Time PCR จะต้องตั้งค่าเกณฑ์สำหรับแต่ละเป้าหมายโดยทั่วไป สัญญาณเรืองแสงของ 15 รอบแรกของปฏิกิริยา PCR จะถูกใช้เป็นสัญญาณพื้นหลังของการเรืองแสง และเกณฑ์การเรืองแสงคือ 10 เท่าของค่าเบี่ยงเบนมาตรฐานของสัญญาณการเรืองแสงของ 3 ถึง 15 รอบแรกของ PCR และเกณฑ์การเรืองแสงถูกตั้งค่าในเฟสเอ็กซ์โปเนนเชียลของการขยาย PCRโดยทั่วไป เครื่องมือแต่ละชนิดมีการตั้งค่าเกณฑ์การเรืองแสงก่อนใช้งาน
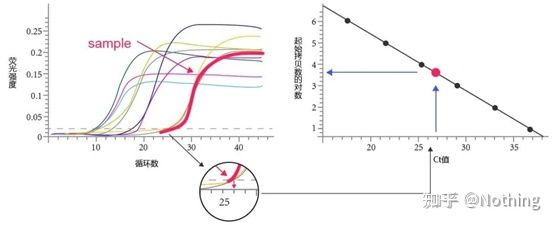
เกณฑ์รอบ (CT) หรือจุดตัด (CP)
รอบที่เส้นโค้งการขยายสัญญาณข้ามเกณฑ์ (เช่น จุดที่การตรวจจับการเรืองแสงเพิ่มขึ้นอย่างมีนัยสำคัญ)CT สามารถเป็นเศษส่วนและสามารถคำนวณจำนวนเทมเพลตเริ่มต้นได้ค่า CT แสดงถึงจำนวนรอบที่พบเมื่อสัญญาณเรืองแสงในหลอดปฏิกิริยา PCR แต่ละหลอดถึงเกณฑ์ที่ตั้งไว้มีความสัมพันธ์เชิงเส้นตรงระหว่างค่า CT ของแต่ละเทมเพลตและลอการิทึมของหมายเลขสำเนาเริ่มต้นของเทมเพลตยิ่งจำนวนสำเนาเริ่มต้นสูง ค่า CT ยิ่งน้อย และในทางกลับกัน.เส้นโค้งมาตรฐานสามารถสร้างขึ้นได้โดยใช้มาตรฐานที่ทราบหมายเลขสำเนาเริ่มต้น โดยที่ abscissa แทนค่า CT และลำดับแทนลอการิทึมของหมายเลขสำเนาเริ่มต้นดังนั้น ตราบเท่าที่ได้รับค่า CT ของตัวอย่างที่ไม่รู้จัก หมายเลขสำเนาเริ่มต้นของตัวอย่างสามารถคำนวณได้จากเส้นโค้งมาตรฐาน
ค่า ΔCT
อธิบายค่า ΔCTความแตกต่างระหว่างยีนเป้าหมายและค่า CT ของยีนอ้างอิงภายในที่สอดคล้องกันเช่น ยีนแม่บ้านทำความสะอาด และใช้เพื่อปรับปริมาณแม่แบบที่ใช้ให้เป็นมาตรฐาน:
⇒ΔCT = CT (ยีนเป้าหมาย) – CT (ยีนอ้างอิงภายนอก)
ค่า ΔΔCT
ค่า ΔΔCT อธิบายความแตกต่างระหว่างค่าเฉลี่ย ΔΔCT ของตัวอย่างที่สนใจ (เช่น เซลล์ที่ถูกกระตุ้น) และค่าเฉลี่ย ΔΔCT ของตัวอย่างอ้างอิง (เช่น เซลล์ที่ไม่ได้กระตุ้น)ตัวอย่างอ้างอิงเรียกอีกอย่างว่าตัวอย่างการสอบเทียบ และตัวอย่างอื่นๆ ทั้งหมดจะถูกทำให้เป็นมาตรฐานสำหรับการวัดปริมาณสัมพัทธ์:
⇒ΔΔCT = เฉลี่ย ΔCT (ตัวอย่างที่สนใจ) – เฉลี่ย ΔCT (ตัวอย่างอ้างอิง)
ยีนอ้างอิงภายนอก (ยีนอ้างอิงภายนอก)
ระดับการแสดงออกของยีนอ้างอิงภายนอก เช่น ยีนดูแลทำความสะอาด (ยีนดูแลทำความสะอาด) ไม่แตกต่างกันระหว่างกลุ่มตัวอย่างการเปรียบเทียบค่า CT ของยีนอ้างอิงกับยีนเป้าหมายทำให้ระดับการแสดงออกของยีนเป้าหมายถูกทำให้เป็นมาตรฐานตามปริมาณของ RNA หรือ cDNA ที่ป้อนเข้า (ดูหัวข้อเกี่ยวกับค่า ΔCT ด้านบน)
ยีนอ้างอิงภายในถูกต้องสำหรับการย่อยสลาย RNA ที่เป็นไปได้หรือการมีอยู่ของสารยับยั้งเอนไซม์ในตัวอย่าง RNA ตลอดจนการแปรผันของเนื้อหา RNA ประสิทธิภาพการถอดรหัสย้อนกลับ การกู้คืนกรดนิวคลีอิก และการจัดการตัวอย่างในการเลือกยีนอ้างอิงที่เหมาะสมที่สุด เราได้แก้ไขอัลกอริทึมเพื่อให้สามารถเลือกยีนอ้างอิงที่เหมาะสมที่สุดโดยขึ้นอยู่กับการตั้งค่าการทดลอง
การควบคุมภายใน
ลำดับการควบคุมที่ขยายในปฏิกิริยาเดียวกันกับลำดับเป้าหมายและโพรบด้วยโพรบอื่น (เช่น ทำ duplex PCR)การควบคุมภายในมักใช้เพื่อตัดการขยายสัญญาณที่ล้มเหลว เช่น เมื่อตรวจไม่พบลำดับเป้าหมาย
ตัวอย่างการสอบเทียบ
ตัวอย่างอ้างอิง (เช่น RNA ที่บริสุทธิ์จากเซลล์หรือเนื้อเยื่อ) ที่ใช้ในการหาปริมาณสัมพัทธ์เพื่อเปรียบเทียบตัวอย่างอื่นๆ ทั้งหมดเพื่อกำหนดระดับการแสดงออกสัมพัทธ์ของยีนตัวอย่างการสอบเทียบสามารถเป็นตัวอย่างใดก็ได้ แต่โดยปกติจะเป็นตัวควบคุม (เช่น ตัวอย่างที่ไม่ผ่านการบำบัดหรือตัวอย่างจากเวลาศูนย์ของการทดลอง)
การควบคุมเชิงบวก
ใช้ปฏิกิริยาควบคุมด้วยจำนวนแม่แบบที่ทราบ.การควบคุมเชิงบวกมักใช้เพื่อตรวจสอบว่าชุดไพรเมอร์หรือชุดไพรเมอร์-โพรบทำงานอย่างถูกต้องและปฏิกิริยาได้รับการตั้งค่าอย่างถูกต้อง
ไม่มีการควบคุมเทมเพลต (กทช)
ปฏิกิริยาควบคุมที่มีองค์ประกอบที่จำเป็นทั้งหมดของปฏิกิริยาการขยาย ยกเว้นแม่แบบ ซึ่งโดยปกติจะถูกแทนที่ด้วยน้ำการใช้ NTC สามารถค้นหาการปนเปื้อนที่เกิดจากการปนเปื้อนของรีเอเจนต์หรือ DNA แปลกปลอม ดังนั้นจึงรับประกันความถูกต้องและความน่าเชื่อถือของข้อมูลการตรวจจับขยายผลคุมกทช.ชี้ปนเปื้อน
ไม่มีการควบคุม RT (NRT)
กระบวนการสกัด RNA อาจมี DNA จีโนมตกค้าง ซึ่งเป็นอันตรายอย่างยิ่งและเป็นตัวการที่ส่งผลต่อคุณภาพของข้อมูลและศัตรูตามธรรมชาติของ qPCR ดังนั้นเมื่อออกแบบการทดลอง จึงต้องออกแบบให้ขยายการตรวจจับ RNA เท่านั้นมีสองวิธี วิธีหนึ่งคือการออกแบบไพรเมอร์ข้ามอินตรอน อีกวิธีคือการกำจัด DNA ออกให้หมด ซึ่งวิธีใดดีกว่า ซึ่งจะกล่าวถึงในภายหลังการควบคุม NTR เป็นกระจกวิเศษในการตรวจจับมลพิษของ DNAหากมีการขยายแสดงว่ามีมลพิษ
มาตรฐาน
มาตรฐานคือตัวอย่างความเข้มข้นที่ทราบหรือหมายเลขสำเนาที่ใช้สร้างเส้นโค้งมาตรฐานเพื่อให้มั่นใจในความเสถียรของมาตรฐาน โดยปกติแล้วชิ้นส่วนของยีนจะถูกโคลนเข้าไปในพลาสมิดและใช้เป็นมาตรฐาน
เส้นโค้งมาตรฐาน
โดยปกติจะเจือจางลงในเกรเดียนต์ความเข้มข้นอย่างน้อย 5 ระดับด้วยผลิตภัณฑ์มาตรฐานตามอัตราส่วนสองเท่า และจุด 5 จุดจะถูกวาดในพิกัดของค่า CT และหมายเลขสำเนา และจุดต่างๆ จะเชื่อมต่อกันเพื่อสร้างเส้นเพื่อสร้างเส้นโค้งมาตรฐานสำหรับเส้นโค้งมาตรฐานแต่ละเส้น จะต้องมีการตรวจสอบความถูกต้องค่าความชันอยู่ระหว่าง –3.3 ถึง –3.8 และแต่ละความเข้มข้นจะดำเนินการเป็นสามเท่าควรทิ้งจุดที่แตกต่างจากจุดอื่นอย่างมีนัยสำคัญค่า CT ของตัวอย่างที่จะทดสอบจะถูกนำเข้ามาในเส้นโค้งมาตรฐาน และสามารถคำนวณระดับการแสดงออกของตัวอย่างที่จะทดสอบได้
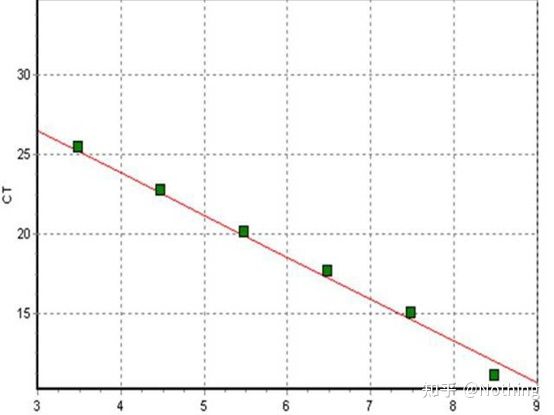
ค่า CT ของตัวอย่างที่จะทดสอบจะนำเข้าสู่เส้นโค้งมาตรฐาน และสามารถคำนวณหมายเลขสำเนาเริ่มต้นของตัวอย่างที่จะทดสอบได้
ประสิทธิภาพและความชัน
ความชันของเส้นโค้งมาตรฐานแสดงถึงประสิทธิภาพของ PCR แบบเรียลไทม์
· ความชัน -3.322 แสดงว่าประสิทธิภาพการขยาย PCR เป็น 1 หรือมีประสิทธิภาพ 100% และปริมาณของผลิตภัณฑ์ PCR เพิ่มขึ้นสองเท่าในแต่ละรอบ
· ความชันน้อยกว่า –3.322 (เช่น –3.8) บ่งชี้ประสิทธิภาพ PCR
· ความชันที่มากกว่า –3.322 (เช่น –3.0) บ่งชี้ว่าประสิทธิภาพ PCR ดูเหมือนจะมากกว่า 100% ซึ่งน่าสงสัยว่า PCR หนึ่งรอบสามารถสร้างผลิตภัณฑ์ที่ขยายได้มากกว่าสองเท่าได้อย่างไรสถานการณ์นี้เกิดขึ้นในเฟสที่ไม่ใช่เชิงเส้นของปฏิกิริยา PCR นั่นคือมีการขยายที่ไม่เฉพาะเจาะจงจำนวนมาก
เส้นโค้งการละลาย
หลังจากการขยาย qPCR เสร็จสิ้น ผลิตภัณฑ์ PCR จะได้รับความร้อนเมื่ออุณหภูมิสูงขึ้น ผลิตภัณฑ์ขยายสัญญาณแบบเกลียวคู่จะค่อยๆ ละลาย ส่งผลให้ความเข้มของแสงเรืองแสงลดลงเมื่อถึงอุณหภูมิที่กำหนด (Tm) ผลิตภัณฑ์จำนวนมากจะละลายการเรืองแสงลดลงอย่างรวดเร็วผลิตภัณฑ์ PCR ที่แตกต่างกันมีค่า Tm ที่แตกต่างกันและอุณหภูมิหลอมเหลวที่แตกต่างกัน เพื่อให้สามารถระบุความจำเพาะของ PCR ได้
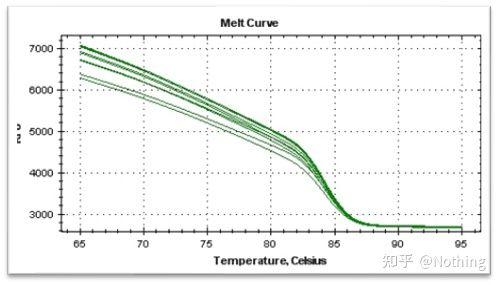
เส้นโค้งการหลอมละลาย (เส้นโค้งอนุพันธ์)
เส้นโค้งการหลอมละลายได้มาจากการสร้างแผนที่สูงสุด ซึ่งสามารถแสดงสถานการณ์ของชิ้นส่วนผลิตภัณฑ์ PCR ได้อย่างเป็นธรรมชาติมากขึ้นเนื่องจากอุณหภูมิหลอมเหลวคือค่า Tm ของชิ้นส่วน DNA จึงสามารถตัดสินพารามิเตอร์บางอย่างที่ส่งผลต่อค่า Tm ของชิ้นส่วน DNA ได้ เช่น ขนาดชิ้นส่วน ปริมาณ GC เป็นต้น โดยทั่วไปแล้ว ตามหลักการออกแบบรองพื้นของเราความยาวของผลิตภัณฑ์ที่ขยายอยู่ในช่วง 80-300bp ดังนั้นอุณหภูมิหลอมเหลวควรอยู่ระหว่าง 80°C ถึง 90°C
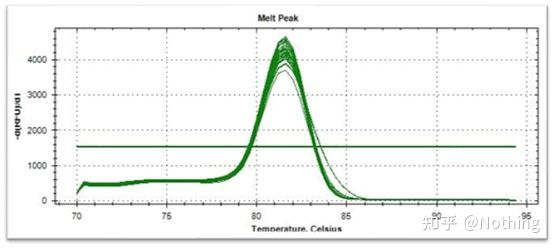
การตีความของเส้นโค้งการละลาย: หากพีคหลักเพียงพีคปรากฏระหว่าง 80°C-90°C แสดงว่า PCR เชิงปริมาณเรืองแสงนั้นสมบูรณ์แบบหากพีคหลักปรากฏขึ้นระหว่าง 80°C-90°C และพีคอื่นปรากฏต่ำกว่า 80°C โดยทั่วไปจะพิจารณาไพรเมอร์ไดเมอร์คุณสามารถลองเพิ่มอุณหภูมิการหลอมเพื่อแก้ปัญหาได้หากยอดหลักปรากฏระหว่าง 80°C-90°C และยอดเบ็ดเตล็ดปรากฏขึ้นอีกครั้งเมื่ออุณหภูมิสูงขึ้น โดยทั่วไปจะถือว่ามีการปนเปื้อนของ DNA และจำเป็นต้องนำ DNA ออกในระยะเริ่มต้นของการทดลอง
แน่นอนว่ายังมีบางสถานการณ์ที่ผิดปกติซึ่งจะแยกย่อยออกไปด้านล่าง
3. ความรู้ขั้นสูง
ในการทำ qPCR ฉันต้องพูดว่า MIQEข้อมูลขั้นต่ำสำหรับการเผยแพร่ของเชิงปริมาณPCR แบบเรียลไทม์การทดลอง—ข้อมูลขั้นต่ำสำหรับการเผยแพร่บทความเกี่ยวกับ PCR เชิงปริมาณตามเวลาจริงการทดลอง .เพื่อให้ทุกคนเข้าใจได้ง่ายขึ้น เราจะลดความซับซ้อนของเนื้อหาหลัก
คุณสามารถค้นหาข้อความต้นฉบับของ MIQE ได้บนอินเทอร์เน็ต และสิ่งที่สำคัญที่สุดคือมันกำหนดเงื่อนไขรายการตรวจสอบข้อมูลที่ต้องมีเมื่อเผยแพร่บทความ .
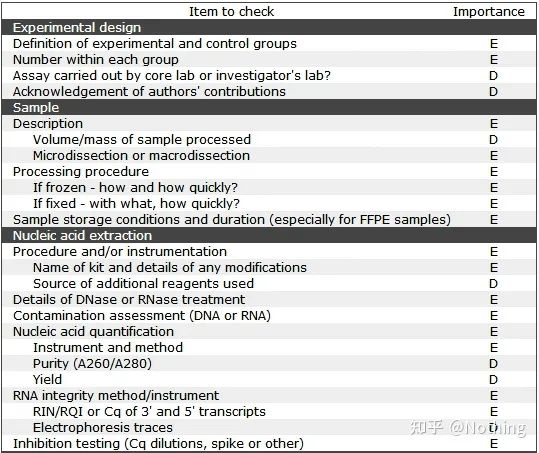
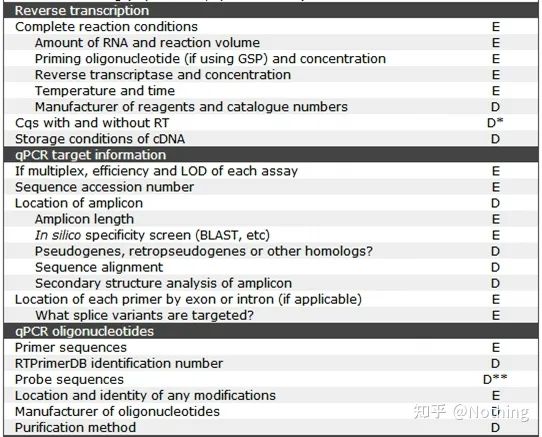
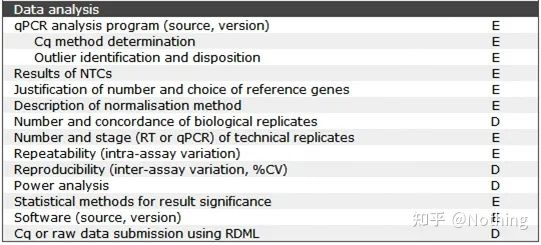
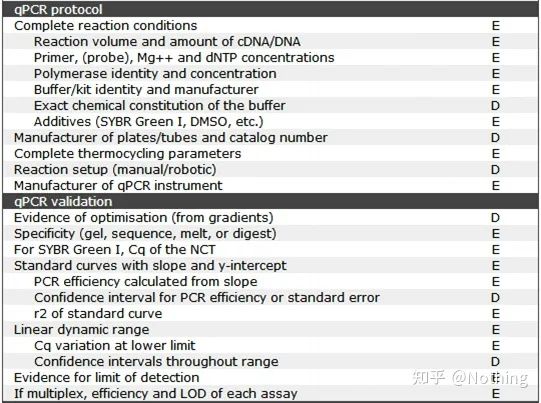
ผู้ตรวจสอบสามารถตัดสินคุณภาพของการทดสอบได้โดยอ่านรายละเอียดเหล่านี้ผู้อ่านในอนาคตสามารถใช้สิ่งนี้เพื่อทำซ้ำหรือปรับปรุงการทดสอบ
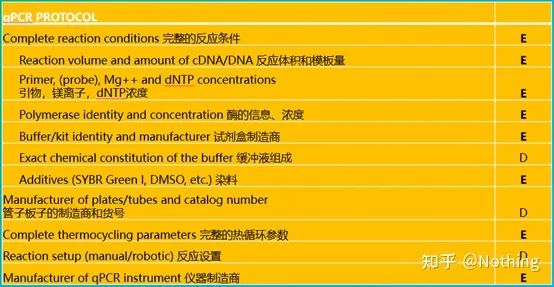
เป็นที่น่าสังเกตว่าในรายการนี้ ความสำคัญของแต่ละรายการจะถูกทำเครื่องหมายด้วย E หรือ D ตามลำดับมันหมายความว่าอะไร?E: ข้อมูลสำคัญ (ต้องส่ง);D: ข้อมูลที่ต้องการ (ให้มากที่สุดเท่าที่จะเป็นไปได้)
MIQE (1)—การออกแบบเชิงทดลอง
พวกสวะหลายคนที่จบวิชาป้องกันตัวหลังจากจบการศึกษาระดับบัณฑิตศึกษาจะไม่รู้วิธีออกแบบการทดลองด้วยตัวเอง เปิดสมุดบันทึก และทำในสิ่งที่อาจารย์บอกให้ทำเป็นผลให้การออกแบบการทดลองไม่เข้มงวดและกองบรรณาธิการของนิตยสารกล่าวว่าพวกเขาต้องการสร้างภาพนี้และภาพนั้น พวกเขาจึงทำด้วยความงุนงงนี่คือวิธีการสร้าง scumbags!

ใกล้บ้านมากขึ้น หลักการแรกของการทดลองคือการพิจารณาความเข้มงวดของตรรกะการทดลอง.สิ่งพื้นฐานที่สุดคือการออกแบบการทดลอง และสิ่งที่สำคัญที่สุดเกี่ยวกับการออกแบบการทดลองคือวิธีตั้งค่าตัวอย่างเป้าหมาย ตัวอย่างอ้างอิง (ตัวควบคุม) และจำนวนการทำซ้ำ เพื่อให้ข้อมูลการทดลองสามารถอ้างอิง เทียบเคียงได้ และน่าเชื่อถือ
ตัวอย่างเป้าหมายหมายถึงตัวอย่างที่ต้องการให้เราตรวจหายีนเป้าหมายหลังจากการรักษาบางอย่างตัวอย่างอ้างอิงเป็นตัวอย่างที่ไม่มีการบำบัดใด ๆ ซึ่งมักเรียกว่าชนิดป่าในทางชีววิทยา
การทดลองซ้ำมีความสำคัญมากโดยทั่วไป จำนวนซ้ำที่โน้มน้าวใจต้องมากกว่าสามจำเป็นต้องแยกแยะว่าการจำลองแบบทางชีวภาพคืออะไรและการจำลองแบบทางเทคนิคคืออะไร
แบบจำลองทางชีวภาพ: การทดลองยืนยันแบบเดียวกันที่ทำกับวัสดุที่แตกต่างกัน (เวลา พืช แบทช์ แผ่นปฏิกิริยา)

การทำสำเนาทางชีวภาพ
ลองใช้การกำจัดศัตรูพืชของพริกไทยเป็นตัวอย่างเราต้องการพ่นสารกำจัดศัตรูพืชบนพืชสามต้นของ ABC จากนั้นพืชทั้งสามของ ABC ก็เป็นแบบจำลองทางชีววิทยาสามต้น และเป็นการทดลองยืนยันแบบเดียวกันที่ดำเนินการโดยใช้วัสดุที่แตกต่างกันแต่ในการทดลอง จำเป็นต้องมีการควบคุมอย่างแน่นอน ดังนั้นเราจึงสามารถฉีดพ่นกิ่งใดกิ่งหนึ่งของพืช A เพื่อสร้างกลุ่มทดลองของพืช A และไม่ฉีดพ่นกิ่งอื่น ๆ ของพืช A เพื่อสร้างกลุ่มควบคุมทำเช่นเดียวกันกับ B และ C
แบบจำลองทางเทคนิค (แบบจำลองทางเทคนิค): เป็นการทดลองซ้ำๆ ที่ออกแบบมาเพื่อหลีกเลี่ยงข้อผิดพลาดที่เกิดจากการทำงาน ซึ่งจริงๆ แล้วรูที่ซ้ำกันรวมอยู่ในวัสดุเดียวกันทั้งการรักษาและการควบคุมต้องมีการตั้งค่าซ้ำ (ขั้นต่ำสาม) ของยีนเป้าหมายและยีนอ้างอิงภายใน
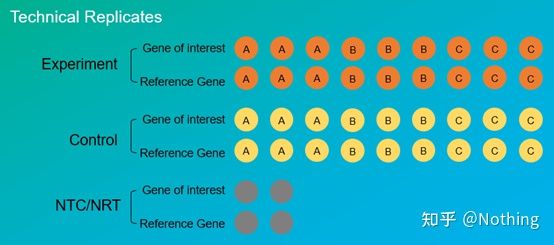
การทำซ้ำทางเทคนิค
ยกตัวอย่างพริกไทยที่ใช้ยาฆ่าแมลงเป็นตัวอย่างอีกครั้งสำหรับกลุ่มทดลองของพืช A เราทำ PCR สามรูที่ 1, 2 และ 3 สำหรับยีนเป้าหมายและยีนอ้างอิงภายในตามลำดับ เพื่อหาค่าเฉลี่ยหลังจากการตรวจจับสำหรับการควบคุมพืช กลุ่ม A ก็ปฏิบัติเช่นเดียวกันทำเช่นเดียวกันกับพืช B และ Cนี่คือการทำซ้ำทางเทคนิค
เป็นที่น่าสังเกตว่าสิ่งที่เข้าสู่สถิติคือการทำซ้ำทางชีววิทยา และการทำซ้ำทางเทคนิคคือการทดสอบว่ามีปรากฏการณ์สุ่มใดๆ ในกระบวนการทดลองหรือไม่ เพื่อให้ผลการทดลองน่าเชื่อถือ นั่นคือเพื่อหลีกเลี่ยงข้อผิดพลาดโดยการหาค่าเฉลี่ยอย่างที่เรามักพูดกัน
การควบคุมเชิงลบ—กทช และ NRT
กทช. (การควบคุมแบบไม่มีเทมเพลต)ใช้ตัวควบคุมที่ไม่มีแม่แบบเพื่อตรวจสอบว่าวัสดุทดลองปนเปื้อนหรือไม่โดยทั่วไปจะใช้น้ำเป็นแม่แบบหากมีปฏิกิริยาเรืองแสง แสดงว่ามีการปนเปื้อนของกรดนิวคลีอิกในห้องปฏิบัติการ
มลพิษเหล่านี้มาจาก: น้ำที่ไม่บริสุทธิ์, รีเอเจนต์ที่ไม่มีคุณภาพซึ่งมี DNA ภายใน, มลพิษจากไพรเมอร์, มลพิษจากอุปกรณ์ในห้องปฏิบัติการ, มลพิษจากละอองลอย ฯลฯ จำเป็นต้องใช้ RNase scavengers และ RNase inhibitorsมลพิษจากละอองลอยเป็นสิ่งที่หาได้ยากที่สุดลองจินตนาการว่าห้องปฏิบัติการของคุณเป็นเหมือนหมอกควันที่มีกรดนิวคลีอิกต่างๆ ลอยอยู่ในอากาศ

NRT (การถอดรหัสแบบไม่ย้อนกลับ)ตัวควบคุมที่ไม่มีการถอดรหัสย้อนกลับ คือ RNA ที่ถอดรหัสแบบไม่ย้อนกลับเป็นตัวควบคุมเชิงลบ ซึ่งเป็นตัวควบคุม gDNA ตกค้าง
เมื่อทำการแสดงออกของยีน ปริมาณของ RNA จะถูกตรวจพบโดยการตรวจหาปริมาณของ cDNA หลังจากการถอดความแบบย้อนกลับหากมี gDNA ตกค้างเมื่อ RNA ถูกทำให้บริสุทธิ์ จะทำให้เกิดความผิดพลาดในผลการทดลอง เนื่องจากผลการทดลองจริงที่ได้คือ gDNA และ cDNAในระดับรวม ไม่ใช่แค่ cDNA เท่านั้น gDNA จำเป็นต้องถูกลบออกทั้งหมดระหว่างการสกัด RNA
MIQE (2)—ข้อมูลตัวอย่าง
ข้อมูลตัวอย่างที่เรียกว่า หมายความว่าเมื่อเราเผยแพร่บทความเกี่ยวกับ qPCR เราต้องอธิบายข้อมูลตัวอย่างอย่างชัดเจน ซึ่งเป็นส่วนที่ขาดไม่ได้ของบทความในทำนองเดียวกัน เมื่อเราดำเนินการกับตัวอย่าง เราต้องควบคุมการดำเนินงานของเราเองด้วยเพื่อให้แน่ใจว่าตัวอย่างถูกต้อง

คำอธิบายของตัวอย่างเป็นเพียงผลลัพธ์เท่านั้น และเราควรให้ความสำคัญกับวัสดุที่นำมาในระหว่างการทดลองทั้งหมด
การเลือกวัสดุการทดลอง
ตัวอย่างเลือด – เลือกเลือดสด ไม่เกิน 4 ชม.ตัวอย่างเซลล์ – เลือกเก็บเซลล์สดในช่วงเวลาที่มีการเติบโตอย่างแข็งแรงเนื้อเยื่อสัตว์—เลือกเนื้อเยื่อที่สดและแข็งแรงเนื้อเยื่อพืช - เลือกเนื้อเยื่อที่สดใหม่
คุณต้องสังเกตว่ามีคำสำคัญในประโยคเหล่านี้: สด
สำหรับตัวอย่างข้างต้น ชุดที่ดีที่สุด คุ้มค่า และเสถียรในตลาดคือชุดของ Foregene ซึ่งสามารถแยก DNA และ RNA ได้อย่างรวดเร็วและง่ายดาย
การจัดเก็บวัสดุการทดลอง
โดยทั่วไป เราไม่แนะนำให้เก็บตัวอย่าง หากเงื่อนไขอนุญาตอย่างไรก็ตาม มีเพื่อนหลายคนที่ไม่สามารถทำการทดลองได้ทันทีหลังจากการสุ่มตัวอย่าง และบางคนถึงกับต้องแบกถังไนโตรเจนเหลวไปที่สนามเพื่อทำการสุ่มตัวอย่าง
สำหรับเพื่อนที่ขยันขันแข็งแบบนี้ ฉันบอกได้คำเดียวว่าคุณไม่เข้าใจวัสดุสิ้นเปลืองของรีเอเจนต์ปัจจุบัน บริษัทที่ใช้รีเอเจนต์จำนวนมากผลิตรีเอเจนต์ที่สามารถเก็บตัวอย่าง RNA ที่อุณหภูมิห้อง และคุณสามารถเลือกใช้ได้วิธีการจัดเก็บแบบเดิมคือการเก็บไนโตรเจนเหลวโดยใช้ถังไนโตรเจนเหลวขนาดเล็กที่พกพาสะดวกหลังจากนำตัวอย่างกลับไปที่ห้องปฏิบัติการแล้ว ให้เก็บไว้ในตู้เย็น -80°C

สำหรับการทดลองเกี่ยวกับ RNA จะต้องปฏิบัติตามหลักการหกคำ:อุณหภูมิต่ำ , ไม่มีเอนไซม์ ,และเร็ว .
แนวคิดเรื่องอุณหภูมิต่ำนั้นเข้าใจง่ายหากไม่มีเอ็นไซม์ RNase จะอยู่ทุกที่ในโลกที่เราอาศัยอยู่ (ไม่เช่นนั้น คุณจะถูก HIV ฆ่าตาย) ดังนั้น วิธีหลีกเลี่ยง RNase เมื่อทำการทดลองจึงเป็นแนวคิดที่สำคัญมากเร็ว,ไม่มีกังฟูใดในโลกที่ทำลายไม่ได้ มีเพียงความเร็วเท่านั้นที่ทำลายไม่ได้.
ดังนั้น ในแง่หนึ่ง ยิ่งเวลาในการสกัดสั้นลงเท่าใด ชุดอุปกรณ์ก็จะยิ่งดีเท่านั้นทำไมฟอร์เจนคิทเน้นความเร็วเพราะรู้ดี
PS: ผู้หญิงบางคนทำการทดลองอย่างระมัดระวัง แต่ก็ไม่ได้ดีเท่าสแลมดังค์หลังจากทำงานมาหลายปีพวกเขารู้สึกว่าพระเจ้าไม่ยุติธรรม บ่นเรื่องคนอื่น และมองหาชีวิตแท้จริงแล้วเธอไม่เข้าใจมันเขาปกป้องอาร์เอ็นเอได้ไม่ดีนัก และผู้เล่นสแลมดังค์ก็ว่องไวตอนที่เขากำลังทำการทดลอง เขาคิดว่าเขาจะจบการสแลมดังก์ด้วยสามครั้ง ห้าครั้ง และสองครั้ง แต่เขาก็ทำการทดลองได้ดี
บันทึก: ช้าลง โอกาสที่ RNase จะบุกรุกมากขึ้นฝึกตัวเองอย่างไรให้รวยเร็ว?ไม่มีทางหรอก แค่ฝึกฝนให้มากขึ้น
สำหรับการทดลองที่แตกต่างกันและตัวอย่างที่แตกต่างกัน ยังคงจำเป็นต้องอ่านเอกสารเพิ่มเติมและเลือกวิธีที่เหมาะสมสำหรับการประมวลผลสำหรับกระบวนการเก็บตัวอย่างและการจัดเก็บ MIQE กำหนดให้ต้องเขียนอย่างชัดเจนในกระดาษ เพื่อให้ผู้ตรวจทานสามารถตรวจสอบความน่าเชื่อถือของกระดาษ และยังสะดวกสำหรับเยาวชนที่มึนงงที่จะทำการทดลองซ้ำ
แม้ว่าการทดลองทางชีววิทยาจะยาก แต่ก็เป็นการทดลองระดับสูงถ้าไม่ระวังก็คว่ำโลกได้ตัวอย่างเช่น การทำให้โรคซาร์สเข้าสู่วิกฤตทางชีวเคมี หรือการทำข้าวลูกผสมเพื่อช่วยชีวิตผู้คน 1.3 พันล้านคนภาพด้านล่างเป็นการทดลองทางเคมี คุณควรเข้าใจว่าคุณภูมิใจในงานวิจัยของคุณแค่ไหนเพียงแค่ดูรูปร่างหน้าตาที่เหมือนจู๋ของเขาลืมมันไปอย่าดำเขา

MIQE (3) – การสกัดกรดนิวคลีอิก
การสกัดกรดนิวคลีอิกเป็นงานใหญ่ และการทดลองทางอณูชีววิทยาทั้งหมดเริ่มต้นด้วยการสกัดกรดนิวคลีอิกก่อนอื่น เรามาคัดลอกเนื้อหาของ MIQE เกี่ยวกับการสกัดกรดนิวคลีอิก
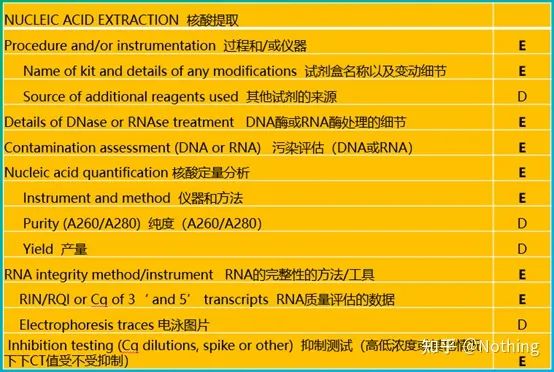
เมื่อมองดูฟอร์มนี้ คุณไม่สามารถอยู่เฉยๆแบบฟอร์มเป็นความเชื่อจะเป็นนักเรียนชั้นนำ คุณต้องถามว่าทำไมเนื้อหาสำคัญของตารางนี้คือ: ติดตามความบริสุทธิ์ ความสมบูรณ์ ความสม่ำเสมอ และปริมาณการสกัดของ RNA .
ส่วนแรกของกระบวนการหรือเครื่องมือคือขั้นตอนการสกัดกรดนิวคลีอิกหากคุณใช้เครื่องสกัดกรดนิวคลีอิกอัตโนมัติเพื่อสกัด (ขั้นสูง โปรดติดต่อฉันเพื่อซื้อ) คุณต้องระบุชื่อรุ่นของเครื่องมือ

ชื่อชุดและ
ชุดเครื่องมือใดที่ใช้สำหรับรายละเอียดการเปลี่ยนแปลง สารรีเอเจนต์พิเศษที่เติมลงไป หรือปฏิบัติการพิเศษใดที่ดำเนินการไปแล้วควรอธิบายให้ชัดเจน เพื่อให้ผู้อื่นสามารถทำการทดลองซ้ำได้อย่างง่ายดาย
บางคนเติมน้ำยาพิเศษเมื่อสกัดตัวอย่างพิเศษ โดยคิดว่านี่คืออาวุธลับของพวกเขาและอย่าบอกคนอื่นในขณะที่เก็บเป็นความลับ พวกเขายังเสียโอกาสในการทำให้บทความของคุณโดดเด่นอีกด้วยอย่าฉลาด คุณต้องซื่อสัตย์มากกว่าประเทศเก่าจางในการวิจัยทางวิทยาศาสตร์ ถ้าคุณอยากฉลาด บทความนี้จะทำให้คุณโง่
ต้องจำหมายเลขผลิตภัณฑ์ของชุดเมื่อคุณสั่งซื้อชุดคิทและเขียนบทความโดยทั่วไปจะมีตัวเลขสองตัวในชุด: Cat—หมายเลขแค็ตตาล็อก (หมายเลขผลิตภัณฑ์ หมายเลขบทความ) ล็อต—หมายเลขล็อตผลิตภัณฑ์ (ใช้เพื่อระบุว่าผลิตภัณฑ์มาจากชุดใด)
นอกจากนี้ หมายเลข CAS มักจะใช้เมื่อสั่งซื้อน้ำยาชีวเคมี และฉันจะทำให้มันเป็นที่นิยมไปพร้อมกันหมายเลข CAS คือหมายเลขที่กำหนดโดย American Chemical Society สำหรับยาเคมีใหม่แต่ละชนิดโดยทั่วไป ตัวเลขสามตัวเชื่อมต่อกันด้วยเส้นประหมายเลข CAS ของ Rushui: 7732-18-5สารเคมีมักมีหลายชื่อแทน แต่หมายเลข CAS นั้นไม่ซ้ำกันในการสั่งซื้อยา สามารถตรวจสอบ CAS Number ของยาก่อนได้
ใกล้บ้าน ทำไมต้องบรรยายเรื่องพวกนี้ให้ชัดเจน?ในความเป็นจริงมันเป็นการตรวจสอบคุณภาพของการสกัด RNAการใช้เครื่องมือและชุดอุปกรณ์จะทำให้การสกัด RNA มีความสม่ำเสมอมากขึ้นระดับการสกัดของห้องปฏิบัติการทั่วไปไม่ใหญ่นักและสามารถรับได้ด้วยชุดอุปกรณ์
รายละเอียดการรักษา DNase หรือ RNase
ประเด็นสำคัญของ fluorescent quantitative PCR คือการป้องกันการปนเปื้อนของ DNA และห้ามทำการทดลองหากมีการปนเปื้อนดังนั้นจึงจำเป็นต้องระบุกระบวนการที่คุณใช้ในการประมวลผล DNA เพื่อแสดงให้เห็นว่า DNA ในกระบวนการทดลองนั้นถูกลบออกอย่างสมบูรณ์และสมบูรณ์แสดงโดยแผนภาพ
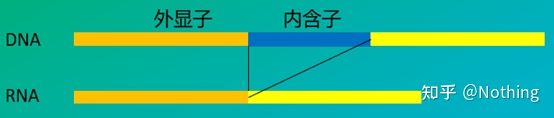
แผนผังของ RNA และ DNA
โดยทั่วไป วิธีการกำจัด DNA คือการรักษา RNA ด้วย DNase หลังจากการสกัดอย่างไรก็ตาม วิธีการเหล่านี้ค่อนข้างเก่าชุดสกัด RNA เชิงพาณิชย์สามารถกำจัด DNA ในระหว่างกระบวนการสกัดโดยไม่ต้องเพิ่ม DNaseตัวอย่างเช่น ชุดอุปกรณ์จาก Foregene
บันทึก: การถอด DNA ออกระหว่างการสกัด RNA เป็นดาบสองคมที่อันตรายมาก ซึ่งจะยืดเวลาการทำงานของการสกัด RNA และเพิ่มความเสี่ยงของการย่อยสลาย RNAโดยพื้นฐานแล้วมันเป็นการแลกเปลี่ยนระหว่างผลผลิต RNA และความบริสุทธิ์
นอกจากนี้ ปริมาณของ DNase ที่เพิ่มลงในคอลัมน์การดูดซับที่มีซิลิกานั้นมีน้อยมาก และต้องใช้ DNase คุณภาพสูงเพื่อให้ได้ผลดังกล่าวไม่สามารถย่อย DNase ที่ไม่เหมาะสมได้อย่างรวดเร็วและสมบูรณ์นี่เป็นการทดสอบระดับทางเทคนิคของผู้ค้าแน่นอนว่ามีพ่อค้าแปลก ๆ ที่โอ้อวดว่าสามารถลบ DNA ออกได้โดยไม่ต้องใช้ DNaseเรียกได้ว่าใครก็ตามที่โม้ว่าสามารถถอด DNA ออกได้หมดโดยไม่มี DNase นั้นเป็นอันธพาลDNA เป็นโครงสร้างแบบเกลียวคู่ที่ค่อนข้างเสถียร และไม่สามารถกำจัดออกได้เพียงแค่พูดคุยและหัวเราะ
การประเมินการปนเปื้อน
วิธีการประเมิน: การตรวจจับด้วยไฟฟ้า, 1% agarose, 6V/cm, 15 นาที, โหลด 1-3 ul
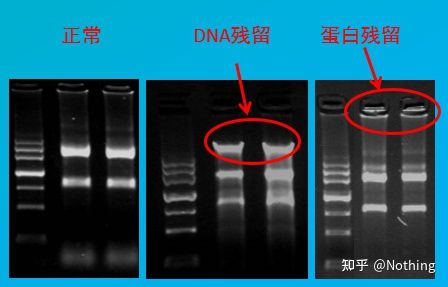
การวิเคราะห์เชิงปริมาณกรดนิวคลีอิก
โดยปกติจะวัดโดยใช้เครื่องสเปกโตรโฟโตมิเตอร์ UVให้ฉันอธิบายความหมายของค่าสามค่าของ OD260, OD280 และ OD230 ก่อน
· OD260nm: เป็นความยาวคลื่นการดูดกลืนของค่าพีคการดูดกลืนสูงสุดของกรดนิวคลีอิก และค่าที่วัดได้ดีที่สุดอยู่ในช่วงตั้งแต่ 0.1 ถึง 1.0หากไม่เป็นเช่นนั้น ให้เจือจางหรือเข้มข้นตัวอย่างเพื่อให้อยู่ในช่วง
·OD280nm: เป็นความยาวคลื่นการดูดกลืนของจุดสูงสุดการดูดกลืนสูงสุดของโปรตีนและสารฟีนอล
· OD230nm: เป็นความยาวคลื่นการดูดกลืนของจุดสูงสุดของการดูดกลืนคาร์โบไฮเดรตสูงสุด
ต่อไปเรามาพูดถึงบทบาทของตัวบ่งชี้แต่ละตัวกันสำหรับ A260 สามารถใช้วัดผลผลิตของกรดนิวคลีอิกได้เมื่อ OD260=1, dsDNA=50μg/ml, ssDNA=37μg/ml, RNA=40μg/ml
เพื่อความบริสุทธิ์ เราต้องดูอัตราส่วนที่เราเห็นกันทั่วไป: OD260/280 และ OD260/230
· DNA บริสุทธิ์: OD260/280 ประมาณเท่ากับ 1.8เมื่อมีค่ามากกว่า 1.9 แสดงว่ามีมลพิษทาง RNA และเมื่อมีค่าน้อยกว่า 1.6 แสดงว่ามีมลพิษทางโปรตีนและฟีนอล
·อาร์เอ็นเอบริสุทธิ์: 1.7
· OD260/230: ไม่ว่าจะเป็น DNA หรือ RNA ค่าอ้างอิงคือ 2.5เมื่อมีค่าน้อยกว่า 2.0 แสดงว่ามีมลพิษของน้ำตาล เกลือ และสารอินทรีย์
ความสมบูรณ์ของอาร์เอ็นเอ
การวัดความสมบูรณ์ของ RNA เป็นสิ่งสำคัญมากโดยทั่วไป จำเป็นต้องทำการทดลอง RNA denaturation gel เพื่อตรวจสอบว่าความสว่างระหว่าง 28S และ 18S RNA มีความสัมพันธ์แบบสองเท่าหรือไม่เมื่อแถบ 5S แถบที่สามปรากฏขึ้น แสดงว่า RNA เริ่มย่อยสลายแล้ว ยกเว้นสัตว์ไม่มีกระดูกสันหลัง

ข้อมูลสำหรับการประเมินคุณภาพ RNA: นอกจากการทดสอบข้างต้นแล้ว ยังมีการทดสอบเครื่องมือขั้นสูงเพิ่มเติมในแง่ของความสมบูรณ์ของ RNA เช่น การทดสอบความสมบูรณ์ของ RQI ของระบบอิเล็กโทรโฟรีซิสอัตโนมัติ Experion ซึ่งสามารถตรวจจับได้ว่า RNA เสื่อมคุณภาพหรือไม่โดยมองไม่เห็น
ในการวิจัยทางวิทยาศาสตร์ fluorescent quantitative PCR คือการเปรียบเทียบระหว่างยีนเป้าหมายกับยีนอ้างอิงภายในดังนั้น ในกระบวนการเก็บรักษาตัวอย่าง RNA การสกัด RNA ฯลฯ เป้าหมายหลักคือเพื่อให้แน่ใจว่า RNA สมบูรณ์
ความสมบูรณ์ของ RNA ส่งผลต่อความสมดุลระหว่างยีนเป้าหมายและยีนอ้างอิงภายในอย่างไร สามารถเข้าใจได้ง่ายจากรูปด้านล่างการย่อยสลายจะนำไปสู่ความไม่สมบูรณ์ของยีน ไม่ว่าจะเป็นความไม่สมบูรณ์ของยีนอ้างอิงภายในหรือความไม่สมบูรณ์ของยีนเป้าหมาย จะมีผลกระทบอย่างมากต่อข้อมูล

แผนผังของยีนเป้าหมายและยีนอ้างอิงต้องไม่เป็นความจริง
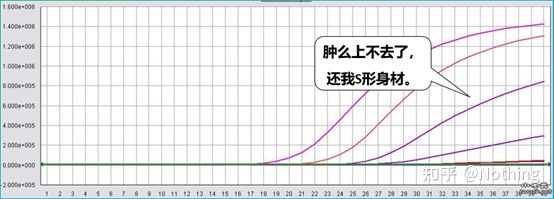
การทดสอบการยับยั้ง (ไม่ว่าค่า CT จะถูกระงับภายใต้ความเข้มข้นสูงหรือต่ำหรือสภาวะอื่นๆ)
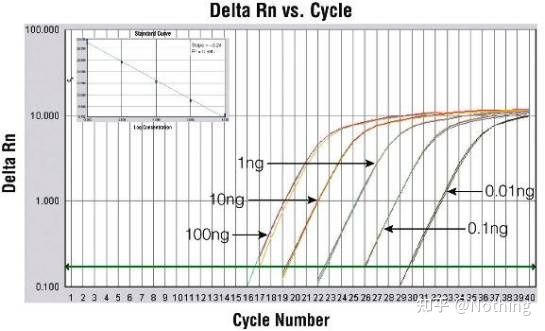
จากตัวเลขนี้เป็นตัวอย่าง ค่า Ct ของเส้นโค้งทั้งห้ามีดังนี้การกระจายของค่า CT ระหว่างเส้นโค้งไม่สม่ำเสมอ และค่า Ct จะล่าช้าภายใต้ความเข้มข้นสูงและต่ำ ซึ่งเป็นกรณีของการยับยั้ง PCR
ประเด็นสำคัญ: ในกระบวนการสกัด RNA เราจำเป็นต้องละทิ้งความเข้าใจผิดและสร้างความเข้าใจที่ถูกต้อง
แนวคิดที่ผิดคือ: การสกัด RNA มุ่งแต่จะให้ผลผลิต โดยคิดว่ายิ่งได้รับ RNA มากเท่าไรก็ยิ่งดีเท่านั้นที่จริงแล้วเมื่อเราทำการหาปริมาณ ถ้าจำนวนยีนไม่มาก เราก็ไม่ต้องการ RNA มากนักปริมาณ RNA ที่คุณดึงออกมามีมากเกินพอ
แนวคิดที่ถูกต้องคือ:การสกัด RNA ควรมีความบริสุทธิ์ ความสมบูรณ์ และความสม่ำเสมอ.ความบริสุทธิ์สามารถรับประกันได้ว่าการถอดรหัสแบบย้อนกลับที่ตามมาจะไม่ถูกยับยั้ง และข้อมูลจะไม่ได้รับผลกระทบจาก DNAความสมบูรณ์ช่วยให้มั่นใจได้ถึงความสมดุลของลำดับเป้าหมายและการอ้างอิงภายในความสม่ำเสมอช่วยให้โหลดตัวอย่างได้เสถียร
MIQE (4) – การถอดความแบบย้อนกลับ
ความเข้าใจผิด: การแสวงหาปริมาณตัวอย่างที่สูงขึ้น
แนวคิดที่ถูกต้อง: ไล่ตามความสม่ำเสมอ (ความเสถียร) โดยไม่คำนึงถึงปริมาณของ RNA ที่โหลด ประสิทธิภาพของการถอดความแบบย้อนกลับยังคงสม่ำเสมอ ทำให้มั่นใจได้ว่าความแตกต่างใน cDNA สามารถสะท้อนถึงความแตกต่างใน mRNA ได้อย่างแท้จริง
เราอธิบายกระบวนการนี้ด้วยแผนภาพ:
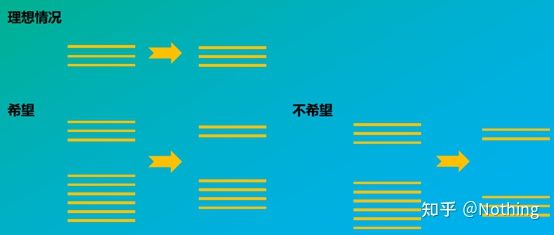
แผนผังของประสิทธิภาพการถอดความย้อนกลับไม่เป็นความจริง
ก่อนอื่น เราต้องเข้าใจความแตกต่างระหว่างกระบวนการถอดความแบบย้อนกลับและกระบวนการ PCRPCR ผ่านกระบวนการให้ความร้อนและการหลอมหลายครั้ง และชิ้นส่วนเป้าหมายจะขยายใหญ่ขึ้นแบบทวีคูณในขณะที่การถอดความแบบย้อนกลับไม่มีกระบวนการนี้ เราสามารถจินตนาการได้ว่าการถอดความแบบย้อนกลับเป็นแบบหนึ่งต่อหนึ่งจริง ๆ ในระหว่างกระบวนการจำลองแบบ เนื่องจาก RNA หลายชิ้น
เนื่องจากสามารถรับข้อมูล cDNA ได้หลายชิ้น ตอนนี้ควรเข้าใจได้แล้ว เนื่องจากชิ้นส่วนขนาดใหญ่และขนาดเล็กได้รับการถอดความแบบย้อนกลับ และเป็นไปไม่ได้ที่จะมุ่งเน้นไปที่ชิ้นส่วนเดียวและเนื่องจากปริมาณของ RNA นั้นค่อนข้างน้อย ปริมาณของ cDNA ที่ได้รับจึงค่อนข้างน้อยเช่นกัน ซึ่งแตกต่างจาก PCR ซึ่งมีผลในการขยาย ดังนั้นจึงเป็นไปไม่ได้ที่จะตรวจจับโดยทั่วไป
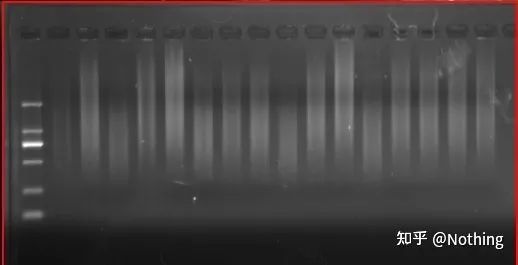
ผลลัพธ์ของ cDNA อิเล็กโตรโฟรีซิส
ประการที่สอง ตามหลักการแล้ว การถอดความแบบย้อนกลับจะดำเนินการแบบหนึ่งต่อหนึ่ง แต่ไม่มีการถอดความแบบย้อนกลับจากบริษัทใด ๆ ที่สามารถบรรลุผลดังกล่าวได้โดยพื้นฐานแล้ว ประสิทธิภาพของรีเวิร์สทรานสคริปเทสส่วนใหญ่จะอยู่ระหว่าง 30-50%หากเป็นกรณีนี้ เราค่อนข้างจะมีประสิทธิภาพการถอดความแบบย้อนกลับค่อนข้างคงที่ ซึ่งเป็นสิ่งที่เราต้องการเห็นในรูป: 3 RNAs รับ 2 cDNAs, 6 RNAs รับ 4 cDNA ดังนั้นไม่ว่าจะโหลดตัวอย่างมากเพียงใด ประสิทธิภาพการถอดความแบบย้อนกลับจะค่อนข้างคงที่เราไม่ต้องการเห็นสถานการณ์ที่ประสิทธิภาพการถอดความย้อนกลับไม่เสถียรและความเข้มข้นสูงถูกยับยั้ง
ดังนั้น จะตรวจสอบได้อย่างไรว่าประสิทธิภาพการถอดความแบบย้อนกลับนั้นเสถียรหรือไม่วิธีการนั้นง่ายมาก คุณเพียงแค่ทำการทดสอบเปรียบเทียบ: วิธีหนึ่งคือการย้อนกลับการถอดเสียงเป็น cDNA หลังจากเจือจาง RNA เป็นสองเท่า และอีกวิธีหนึ่งคือการทำให้การเจือจางเป็นสองเท่าหลังจากย้อนกลับการถอดเสียงเป็น cDNA จากนั้นทำ qPCR เพื่อดูว่าความชันที่ได้นั้นสอดคล้องกันหรือไม่ในฐานะนักเรียนชั้นนำ คุณควรเข้าใจมันในไม่กี่วินาทีดังที่แสดงด้านล่าง:
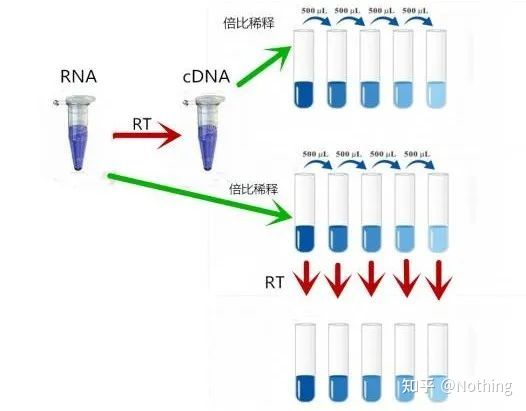
การเจือจาง RNA และ cDNA เพื่อทดสอบว่าประสิทธิภาพของการถอดรหัสแบบย้อนกลับนั้นเสถียรหรือไม่
ทรานสคริปเทสย้อนกลับและคิท
PCR เชิงปริมาณฟลูออเรสเซนต์ที่สมบูรณ์แบบจะมี reverse transcriptase และ kit ที่ยอดเยี่ยมได้อย่างไรReverse transcriptase แบ่งออกเป็นสองประเภทตามแหล่งที่มาอย่างคร่าว ๆ คือ AMV หรือM-MLVและประสิทธิภาพจะเหมือนกับที่แสดงในตาราง
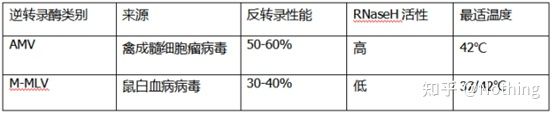
กิจกรรม RNase H
RNase H คือ Ribonuclease H ชื่อภาษาจีนคือ ribonuclease H ซึ่งเป็น endoribonuclease ที่สามารถไฮโดรไลซ์ RNA ในสายโซ่ลูกผสม DNA-RNA ได้RNase H ไม่สามารถไฮโดรไลซ์พันธะฟอสโฟไดเอสเทอร์ใน DNA หรือ RNA แบบสายเดี่ยวหรือสายคู่ได้ นั่นคือ ไม่สามารถย่อย DNA หรือ RNA สายเดี่ยวหรือสายคู่ได้มักใช้ในการสังเคราะห์ cDNA สายที่สอง
มันเป็นเรื่องแปลกเราบอกว่ารีเวิร์สทรานสคริปเทสมีกิจกรรม RNase H ไม่ใช่ว่ารีเวิร์สทรานสคริปเทสมี RNase H และอาจเป็นไปไม่ได้ที่จะแยก RNase H ออกจากรีเวิร์สทรานสคริปเทส อาจเป็นเพราะโครงสร้างของกลุ่มบางกลุ่มในรีเวิร์สทรานสคริปเทส กิจกรรมนี้เกิดจากรีเวิร์สทรานสคริปเทส
ดังนั้น โดยไม่คำนึงถึงประสิทธิภาพการถอดรหัสแบบย้อนกลับของ AMV ที่สูงกว่า กิจกรรมของ RNase H ของมันจะลดผลผลิตของ cDNAแน่นอน ผู้ผลิตรีเอเจนต์กำลังเพิ่มประสิทธิภาพผลิตภัณฑ์ของตนอย่างต่อเนื่องเพื่อกำจัดกิจกรรมของ RNase H ในรีเวิร์สทรานสคริปเทสให้มากที่สุดเท่าที่จะเป็นไปได้เพื่อเพิ่มผลผลิตของ cDNA
อุณหภูมิการหลอม
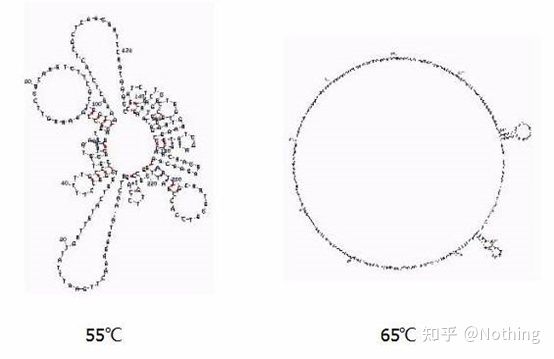
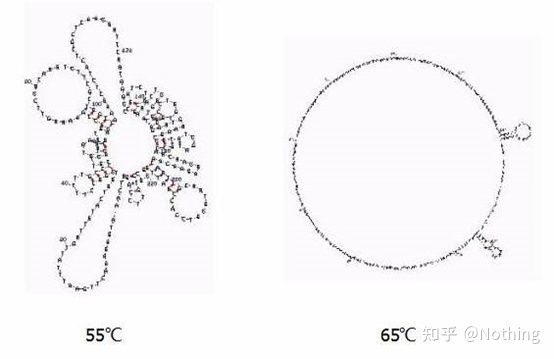
โครงสร้างทุติยภูมิของ RNA ที่อุณหภูมิต่างกัน
ดูภาพด้านบนสำหรับโครงสร้างทุติยภูมิของ RNA ที่อุณหภูมิต่างๆ และใช้เครื่องมือออนไลน์ mFold เพื่อกำหนดโครงสร้างทุติยภูมิของชิ้นส่วนเป้าหมายภายใต้สภาวะอุณหภูมิและความเข้มข้นของเกลือที่เฉพาะเจาะจงที่อุณหภูมิ 55°C โครงสร้างทุติยภูมิของ RNA ยังคงซับซ้อนมาก รีเวิร์สทรานสคริปเทสไม่สามารถทำงานได้ และโครงสร้างทุติยภูมิไม่สามารถแก้ไขได้อย่างสมบูรณ์จนกว่าจะถึง 65°C ในขณะที่อุณหภูมิที่เหมาะสมของ AMV และ M-MLV นั้นต่ำกว่าอุณหภูมินี้มาก
จะทำอย่างไร?โครงสร้างทุติยภูมิคือการจับคู่เสริมของเทมเพลตเอง ซึ่งนำไปสู่การแข่งขันที่รุนแรงระหว่างไพรเมอร์และรีเวิร์สทรานสคริปเทสกับเทมเพลต ส่งผลให้เกิดปัญหาต่างๆ เช่น ค่า E ต่ำและการทำซ้ำได้ไม่ดี
จะทำอย่างไร?เพิ่มอุณหภูมิการหลอมให้มากที่สุดเท่านั้น
ผู้ผลิตรีเอเจนต์หลายรายกำลังปรับปรุงรีเวิร์สทรานสคริปเทสผ่านทางพันธุวิศวกรรมบางชนิดเพิ่มอุณหภูมิของปฏิกิริยา เช่น Jifan และ Aidelai และบางชนิดก็กำจัดกลุ่มที่ใช้งานของเอนไซม์ RNase H เพื่อปรับปรุงความสัมพันธ์ระหว่างเอนไซม์และแม่แบบ RNAความสัมพันธ์สูงสามารถแข่งขันบีบโครงสร้างรองและอ่านผ่านได้อย่างราบรื่น และยังปรับปรุงประสิทธิภาพของการถอดความแบบย้อนกลับได้อย่างมาก
ประเด็นสำคัญ: การถอดความแบบย้อนกลับมีความสำคัญมากกว่าในการติดตามความสม่ำเสมอของประสิทธิภาพการถอดความแบบย้อนกลับ (เอนไซม์ต้องไม่เพียงมีประสิทธิภาพ แต่ยังมีความเสถียรด้วย) มากกว่าจำนวนตัวอย่างที่โหลด หากไม่ใช่ PCR เชิงปริมาณฟลูออเรสเซนต์ขนาดใหญ่เป็นพิเศษ ก็จะไม่สามารถทำได้เลยcDNA หลายตัว
ผู้ผลิตหลายรายได้พยายามแสวงหาความสอดคล้องกันตัวอย่างเช่น บริษัทส่วนใหญ่ได้บรรจุการถอดความแบบย้อนกลับเป็นชุดมาตรฐานสำหรับขาย ซึ่งเป็นทางเลือกที่ดี
ตัวอย่างเช่น ชุดอุปกรณ์ RT Easy Series ของ Foregene:
RT Easy I (มาสเตอร์พรีมิกซ์สำหรับชุดการสังเคราะห์ cDNA สายแรก)
MIQE (5) – ข้อมูลยีนเป้าหมาย

รูปด้านบนอธิบาย
1. ไม่ว่ายีนนี้จะมีประสิทธิภาพสำหรับการทดลองซ้ำหรือไม่นั้นสามารถยืนยันได้โดยการทดลองซ้ำ
2. Gene ID คุณรู้
3. ความยาวยีน ความยาวรวมของยีนเป้าหมายไม่มีปัญหาแน่นอนเมื่อออกแบบไพรเมอร์ ตรวจสอบให้แน่ใจว่าความยาวของแอมพลิคอนอยู่ระหว่าง 80-200bp เพื่อให้แน่ใจว่าประสิทธิภาพการขยายเสียงดีขึ้น
4. ข้อมูลการเปรียบเทียบ Sequence Blast จำเป็นต้องเปรียบเทียบยีนเป้าหมายในธนาคารยีนเพื่อป้องกันการขยายที่ไม่เฉพาะเจาะจง
5. การปรากฏตัวของ pseudogenespseudogene เป็นลำดับดีเอ็นเอที่คล้ายกับยีนปกติ แต่สูญเสียหน้าที่ปกติไปมักอยู่ในตระกูลยูคาริโอตหลายยีนโดยปกติจะแสดงด้วย ψเป็นสำเนาดีเอ็นเอของจีโนมที่ไม่สามารถใช้งานได้ในจีโนมซึ่งคล้ายกับลำดับของยีนที่เข้ารหัสโดยทั่วไปจะไม่ถอดความ และไม่มีความหมายทางสรีรวิทยาที่ชัดเจน
6. ตำแหน่งของไพรเมอร์เทียบกับ exons และ intronsในช่วงปีแรก ๆ เมื่อเราแก้ปัญหาการปนเปื้อนของ DNA เรามักจะให้ความสนใจกับตำแหน่งของไพรเมอร์ เอ็กซอน และอินตรอน และโดยทั่วไปแล้วถือว่าการออกแบบไพรเมอร์ข้ามอินตรอนเพื่อหลีกเลี่ยงการขยายของดีเอ็นเอโปรดดูรูปด้านล่าง: สีดำแทนอินตรอน สีฟ้าต่างๆ แทนเอ็กซอน สีชมพูแทนไพรเมอร์ทั่วไป และสีแดงสดแทนไพรเมอร์ช่วงอินตรอน
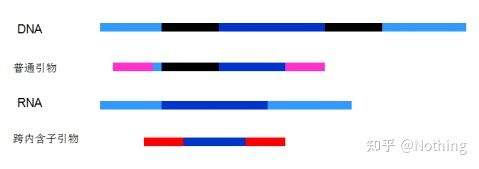
แผนผังไม่เคยเป็นจริง
ช่างเป็นแผนที่สมบูรณ์แบบอะไรเช่นนี้ แต่จริงๆ แล้ว ในกรณีส่วนใหญ่ ไพรเมอร์ทรานส์อินตรอนไม่ได้วิเศษอย่างที่คิด และพวกมันยังทำให้เกิดการขยายที่ไม่เฉพาะเจาะจงอีกด้วยดังนั้นวิธีที่ดีที่สุดในการป้องกันการปนเปื้อนของ DNA คือการกำจัด DNA ออกให้หมด
7. การทำนายโครงสร้างในตัวอย่างนี้อีกครั้ง ใช้เครื่องมือออนไลน์ mFold เพื่อกำหนดโครงสร้างรองของชิ้นส่วนเป้าหมายที่อุณหภูมิและความเข้มข้นของเกลือเฉพาะ
โครงสร้างทุติยภูมิของ RNA ที่อุณหภูมิต่างกัน
โครงสร้างรองคือการจับคู่เสริมของเทมเพลตเอง ซึ่งจะนำไปสู่การแข่งขันที่รุนแรงระหว่างการจับคู่ไพรเมอร์และเทมเพลต และโอกาสของการรวมไพรเมอร์ก็น้อยลง ส่งผลให้เกิดปัญหาหลายอย่าง เช่น ค่า E ต่ำและการทำซ้ำได้ไม่ดีผ่านการคาดคะเนซอฟต์แวร์ ถ้าไม่มีปัญหาโครงสร้างรอง นั่นจะดีมากหากมี บทความติดตามผลของเราจะกล่าวถึงวิธีแก้ปัญหานี้โดยเฉพาะ
MIQE (6)—qPCR โอลิโกนิวคลีโอไทด์

สำหรับ PCR เชิงปริมาณฟลูออเรสเซนต์ สิ่งแรกที่คุณต้องเผชิญทุกวันคือการสกัด RNA และสิ่งที่สองอาจเป็นการออกแบบไพรเมอร์
ก่อนอื่น เรายังคงตรวจสอบกฎเกี่ยวกับการออกแบบสีรองพื้นตามรายการตรวจสอบ MIQEมันง่ายมากจนพวกขี้โกงหัวเราะได้ และเราสามารถจบมันได้ในประโยคเดียว: ค้นหาลำดับและตำแหน่งของไพรเมอร์โพรบ และวิธีการดัดแปลงสำหรับวิธีการทำให้บริสุทธิ์ด้วยไพรเมอร์ ปัจจุบันการสังเคราะห์ไพรเมอร์มีราคาถูกมาก qPCR นั้นคู่ควรกับ PAGE และเหนือกว่าวิธีการทำให้บริสุทธิ์ และข้อมูลของเครื่องมือสังเคราะห์นั้นไม่สำคัญหลายคนทำไพรเมอร์มาหลายทศวรรษแล้วและไม่รู้ว่าซินธิไซเซอร์คือ ABI3900
เกี่ยวกับหลักการของการออกแบบไพรเมอร์ คุณไม่จำเป็นต้องท่องจำเพราะซอฟต์แวร์ออกแบบไพรเมอร์หรือเครื่องมือออนไลน์ส่วนใหญ่สามารถจัดการปัญหาเหล่านี้ได้ (เครื่องมือออนไลน์ที่แนะนำ primer3.ut.ee/) และ 99.999% ของการออกแบบไพรเมอร์ไม่ได้ทำด้วยมือ ดูสิ ผู้เขียนบางครั้งออกแบบไพรเมอร์เป็นร้อยตัวต่อวัน ถ้าคุณอ่านทีละตัว มันก็จะไขว้เขว
เพียงตรวจสอบประเด็นต่อไปนี้หลังจากออกแบบไพรเมอร์แล้ว:
1. ออกแบบไพรเมอร์ใกล้กับปลาย 3′: ในกรณีของการใช้ไพรเมอร์ oligo dT สำหรับการสังเคราะห์ cDNA สายแรก โดยพิจารณาจากประสิทธิภาพการถอดรหัสย้อนกลับและความสมบูรณ์ของ RNA ไพรเมอร์ที่ออกแบบจำเป็นต้องออกแบบใกล้กับปลาย 3′ เพื่อปรับปรุงประสิทธิภาพการขยายสัญญาณใช้รูปอธิบายดังนี้(ไม่มีทางเข้าใจหรอก)
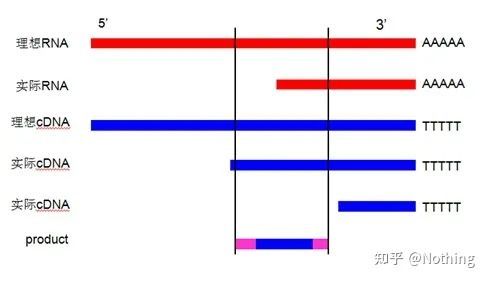
ทำไมไพรเมอร์จึงควรออกแบบให้ใกล้กับปลาย 3′ นั้นจะต้องไม่เป็นความจริง
2. ค่า TM: ค่า Tm อยู่ที่ 55-65°C (เนื่องจากกิจกรรม exonuclease สูงสุดที่ 60°C) และเนื้อหา GC อยู่ที่ 40%-60%
3. BLAST: เพื่อหลีกเลี่ยงการขยายจีโนมที่ไม่เฉพาะเจาะจง จึงต้องใช้ Blast สำหรับการตรวจสอบเพิ่มเติม
MIQE(7)—กระบวนการ qPCR
1. ชุด qPCR
ตามข้อกำหนดของ MIQE เราต้องอธิบายเงื่อนไขการเกิดปฏิกิริยาทั้งหมดอย่างชัดเจนในบทความ รวมถึงการกำหนดค่าของระบบปฏิกิริยา PCR ชุดอุปกรณ์ที่ใช้ ใครคือผู้ผลิต ระบบปฏิกิริยามีขนาดใหญ่เพียงใด ไม่ว่าจะใช้วิธีย้อมหรือวิธีโพรบ การตั้งค่าโปรแกรม PCRนักขับรุ่นเก๋าจะพบว่าตราบเท่าที่มีการเลือกชุดอุปกรณ์ ข้อมูลข้างต้นจะถูกกำหนดโดยพื้นฐานแล้ว
ในปัจจุบัน การผลิตและการผลิตชุด PCR เชิงปริมาณเรืองแสงเป็นเทคโนโลยีที่เติบโตเต็มที่ตราบใดที่คุณไม่เลือกผู้ผลิตที่ห่วยแตกมาก ความน่าจะเป็นของปัญหาก็ไม่สูงนัก แต่เรายังต้องการแบ่งปันบางประเด็นกับคุณ:
เอนไซม์ Taq ที่เริ่มร้อน:ส่วนที่สำคัญที่สุดของ PCR คือเอนไซม์ Taq ที่เริ่มร้อนเอ็นไซม์ hot-start ในตลาดโดยทั่วไปแบ่งออกเป็น 2 ประเภท ประเภทหนึ่งคือเอนไซม์ hot-start ที่ดัดแปลงทางเคมี (คุณสามารถจินตนาการได้ว่าเป็นการฝังพาราฟิน) และอีกประเภทคือ เป็นเอนไซม์ hot-start สำหรับการปรับเปลี่ยนแอนติบอดี (แอนติเจน-แอนติบอดีจับ)การดัดแปลงทางเคมีเป็นวิธีเริ่มต้นของเอนไซม์ที่เริ่มร้อนเมื่อถึงอุณหภูมิที่กำหนด เอ็นไซม์จะปล่อยกิจกรรมออกมาเอนไซม์ hot-start ที่ดัดแปลงแอนติบอดีใช้วิธีการทางชีววิทยาเพื่อขัดขวางการทำงานของเอนไซม์เมื่อถึงอุณหภูมิที่กำหนด แอนติบอดีจะถูกทำให้เสียสภาพธรรมชาติและถูกปิดการใช้งานในฐานะโปรตีน และกิจกรรมของเอนไซม์จะถูกนำมาใช้
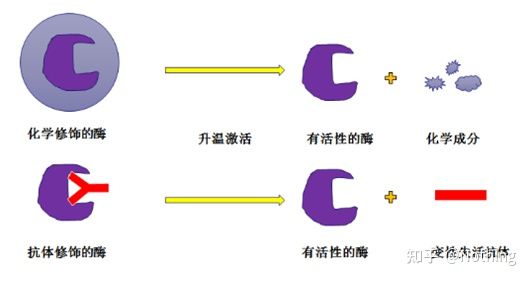
อย่างไรก็ตามสิ่งนี้มีประโยชน์อย่างไรในกรณีนี้ กิจกรรมการปลดปล่อยของเอนไซม์ที่ดัดแปลงแอนติบอดีนั้นเร็วกว่าของเอนไซม์ที่ดัดแปลงทางเคมี ดังนั้นในแง่ของความไว เอนไซม์ที่ดัดแปลงแอนติบอดีมีข้อได้เปรียบเล็กน้อย ดังนั้นจึงไม่มีเอนไซม์ที่ดัดแปลงทางเคมีโดยพื้นฐานในชุดอุปกรณ์ในตลาดหากมีแสดงว่าเทคโนโลยีของผู้ผลิตรายนี้ยังคงติดอยู่ในยุคสหัสวรรษ
ความเข้มข้นของแมกนีเซียมไอออน:ความเข้มข้นของแมกนีเซียมไอออนมีความสำคัญมากในปฏิกิริยา PCRความเข้มข้นของแมกนีเซียมไอออนที่เหมาะสมสามารถส่งเสริมการปลดปล่อยกิจกรรมของเอนไซม์ Taqหากความเข้มข้นต่ำเกินไป กิจกรรมของเอนไซม์จะลดลงอย่างมากหากความเข้มข้นสูงเกินไป การขยายแบบไม่เฉพาะเจาะจงที่เร่งปฏิกิริยาด้วยเอนไซม์จะเพิ่มขึ้นความเข้มข้นของแมกนีเซียมไอออนจะส่งผลต่อการหลอมไพรเมอร์ อุณหภูมิหลอมเหลวของแม่แบบและผลิตภัณฑ์ PCR ซึ่งจะส่งผลต่อผลผลิตของชิ้นส่วนขยายโดยทั่วไปความเข้มข้นของไอออนแมกนีเซียมจะถูกควบคุมที่ 25mMแน่นอน สำหรับชุดอุปกรณ์ที่ดี ความเข้มข้นของไอออนแมกนีเซียมจะต้องได้รับการควบคุมอย่างดีผู้ค้าบางรายเพิ่มสารคีเลตแมกนีเซียมไอออนลงในรีเอเจนต์ ซึ่งสามารถบรรลุผลของการปรับความเข้มข้นของแมกนีเซียมไอออนโดยอัตโนมัติ
ความเข้มข้นของสีย้อมเรืองแสง:สีย้อมเรืองแสงซึ่งเป็นสีเขียว SYBR ที่เรามักจะใช้ ส่วนใหญ่สร้างสารเรืองแสงโดยการจับกับร่องเล็กๆ ของ DNA สายคู่ เนื่องจากการจับสีย้อมกับ DNA สายคู่นั้นไม่เฉพาะเจาะจง กล่าวคือ ตราบใดที่ DNA สายคู่ถูกรวมเข้าด้วยกัน การเรืองแสงสามารถเกิดขึ้นได้ ดังนั้นไพรเมอร์-ไดเมอร์และแม่แบบ DNA ในระบบจะรวมเข้าด้วยกันเพื่อสร้างสัญญาณพื้นหลัง
PS: เนื่องจากคุณสมบัติที่ไวต่อแสง ผลิตภัณฑ์ในท้องตลาดมักบรรจุในหลอดหมุนเหวี่ยงสีขาวขุ่นสีน้ำตาล (ตามภาพด้านล่าง)อย่างไรก็ตามสิ่งนี้จะพบปัญหาเป็นการยากที่จะดูว่าของเหลวถูกดูดหรือไม่เมื่อสุ่มตัวอย่างในแง่นี้ Qingke เป็นมิตรกับผู้ใช้มากที่สุด (ดังแสดงในภาพด้านล่าง) และหลอดใสบรรจุในถุงดีบุกทึบแสงจากนั้นใส่ลงในถุงดีบุกโดยคำนึงถึงความสะดวกในการหลีกเลี่ยงแสงและการสุ่มตัวอย่างคุณต้องเลือกหมายเลขผลิตภัณฑ์ที่ถูกต้องTSE204 เป็นสิ่งมีชีวิตที่คุ้มค่ามาก ทำให้ฉันอยากปลูกหญ้า

ความเข้มข้นของสีย้อมเรืองแสงก็มีความสำคัญเช่นกันหากความเข้มข้นต่ำเกินไป เส้นโค้งการขยายจะไม่เพิ่มขึ้นในระยะหลังและไม่สมบูรณ์หากความเข้มข้นสูงเกินไปจะทำให้เกิดสัญญาณรบกวนเนื่องจาก PCR เชิงปริมาณของฟลูออเรสเซนต์ขึ้นอยู่กับค่า CT เป็นหลัก หากไม่ได้ปรับความเข้มข้นของสีย้อมเรืองแสงอย่างเหมาะสม จุดต่ำสุดจะดีกว่าจุดสูงแน่นอนว่าความเข้มข้นของสีย้อมที่เหมาะสมนั้นดีที่สุด
ROX: สีย้อม ROX ใช้เพื่อแก้ไขข้อผิดพลาดของสัญญาณเรืองแสงจากหลุมถึงหลุมผู้ผลิตเครื่องมือบางรายต้องการการสอบเทียบ ในขณะที่ผู้ผลิตรายอื่นไม่ต้องการตัวอย่างเช่น การใช้เครื่องขยายสัญญาณ Real Time PCR ของ Thermo Fisher Scientific มักจะต้องมีการสอบเทียบ รวมถึง 7300, 7500, 7500Fast, StepOnePlus เป็นต้น คำแนะนำชุดทั่วไปจะอธิบายไว้
qPCR Mix ของ Foregene ยังมีสีย้อม ROX ซึ่งสะดวกสำหรับการใช้งานในรุ่นต่างๆ
การรักษาพันธะไฮโดรเจนอย่างอ่อน: การรักษาพันธะไฮโดรเจนที่อ่อนแอเป็นเรื่องทางเทคนิคไม่มีอะไรอ่านคู่มือของชุดอุปกรณ์จำนวนมาก แต่ไม่มีใครพูดถึงหัวข้อนี้ในความเป็นจริงมันสำคัญมากการรวมกันของเบสส่วนใหญ่ขึ้นอยู่กับความแข็งแรงของพันธะไฮโดรเจนพันธะไฮโดรเจนที่แข็งแรงคือการขยายตัวตามปกติ และพันธะไฮโดรเจนที่อ่อนแอจะนำไปสู่การขยายที่ไม่เฉพาะเจาะจงหากไม่สามารถกำจัดพันธะไฮโดรเจนที่อ่อนแอได้ดี จะไม่สามารถหลีกเลี่ยงการขยายแบบไม่เฉพาะเจาะจงได้ภายในขอบเขตของผู้เขียน มีเพียงไม่กี่บริษัทเท่านั้นที่สังเกตเห็นปัญหานี้เมื่อคุณซื้อชุดอุปกรณ์ คุณสามารถอ้างอิงได้ว่าคุณได้พิจารณาวิธีแก้ปัญหาในเรื่องนี้สำหรับชุดอุปกรณ์ที่คุณต้องการเลือกหรือไม่
ปริมาณปฏิกิริยา: ระบบ 20-50ul เป็นที่นิยมใช้มากกว่า และไดรฟ์ข้อมูลขนาดเล็กมีแนวโน้มที่จะทำให้เกิดข้อผิดพลาดโดยทั่วไป คำแนะนำชุดเครื่องมือจะแนะนำให้ใช้ปริมาณปฏิกิริยา PCRอย่าฉลาดและใช้ปริมาณที่น้อยลงเพื่อประหยัดค่าใช้จ่ายเป้าหมายของ.ปริมาณที่แนะนำโดยผู้ค้าได้รับการทดสอบจริงแล้ว และอาจเป็นไปได้ว่าพวกเขาไม่สามารถแก้ปัญหาข้อผิดพลาดที่เกิดจากปริมาณน้อยได้
2. ผู้ผลิตและหมายเลขบทความของแผ่นท่อ
ทุกคนรู้หลักการของ PCR เชิงปริมาณเรืองแสงการเก็บฟลูออเรสเซนต์ส่วนใหญ่ดำเนินการผ่านฝาท่อ PCRเมื่อเลือกวัสดุสิ้นเปลือง PCR ให้ใส่ใจกับสองจุด: การส่งผ่านแสงที่ดีและเหมาะสมกับเครื่องมือโดยทั่วไปแล้ว บอร์ดและท่อของแบรนด์ทั่วไปนั้นใช้ได้ แต่คุณต้องเลือกอย่างรอบคอบในแง่ของการปรับตัว มิฉะนั้น คุณจะไม่สามารถใช้เครื่องดนตรีได้

4. ความรู้ระดับสูง
MIQE (8)—การตรวจสอบ qPCR
นี่คือความสำคัญสูงสุดของ qPCR!วีรบุรุษมากมายได้ล้มลงบนผืนทรายที่นี่แน่นอน เป็นไปได้ว่าคุณโชคดีและยีนที่คุณศึกษานั้นเรียบง่าย ดังนั้นคุณจึงลอยผ่านถ้ำน้ำแข็งไปตามสายลมข้อมูลการตรวจสอบของ qPCR มีวัตถุประสงค์เพื่อทดสอบความน่าเชื่อถือของข้อมูลเราแสดงรายการข้อมูลการยืนยันที่จำเป็นดังต่อไปนี้:
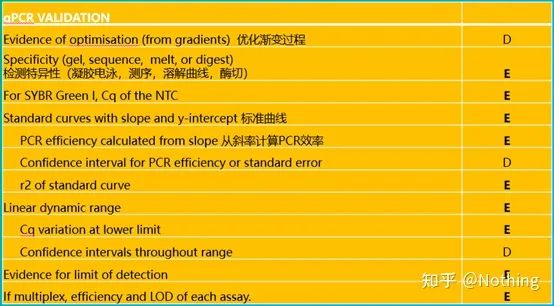
1. การทดสอบความเฉพาะเจาะจง
ความจำเพาะของการขยายยีนเป้าหมายได้รับการทดสอบโดยตรวจสอบว่าภาพอิเล็กโตรโฟรีซิสเป็นแถบเดียวหรือไม่การตรวจสอบลำดับ;เส้นโค้งละลายเพื่อดูว่าแผนที่จุดสูงสุดเป็นแบบเดี่ยวหรือไม่การตรวจสอบการย่อยด้วยเอนไซม์และวิธีอื่นๆ
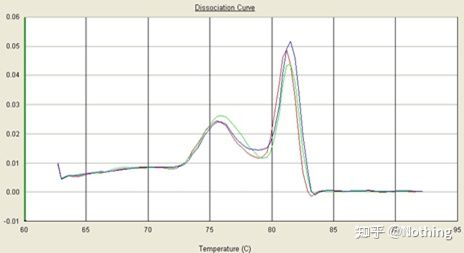
ที่นี่เรามุ่งเน้นไปที่ tเขาวิเคราะห์การขยายแบบไม่เฉพาะเจาะจงโดยใช้วิธีการละลายเส้นโค้ง.โดยทั่วไปแล้ว เมื่อเราออกแบบไพรเมอร์ ขนาดของชิ้นส่วนผลิตภัณฑ์จะต้องอยู่ในช่วง 80-200bp ซึ่งทำให้อุณหภูมิหลอมเหลวของผลิตภัณฑ์ PCR อยู่ในช่วง 80-85 °Cดังนั้น หากมีพีคเบ็ดเตล็ด จะต้องมีผลิตภัณฑ์ขยายเสียงอื่นๆ ที่ไม่เฉพาะเจาะจงหากจุดสูงสุดปรากฏต่ำกว่า 80°C โดยทั่วไปจะถือว่าเป็นไพรเมอร์ไดเมอร์หากจุดสูงสุดปรากฏสูงกว่า 85°C โดยทั่วไปจะถือว่าเป็นการปนเปื้อนของ DNA หรือมากกว่านั้น การขยายแบบไม่เฉพาะเจาะจงของชิ้นส่วนขนาดใหญ่
หมายเหตุ: บางครั้งจะมีพีคเดียวที่ 80°Cในเวลานี้ต้องยึดแนวคิดนี้เป็นไปได้ว่าผลลัพธ์ของการขยายเสียงคือไพรเมอร์ไดเมอร์ทั้งหมด
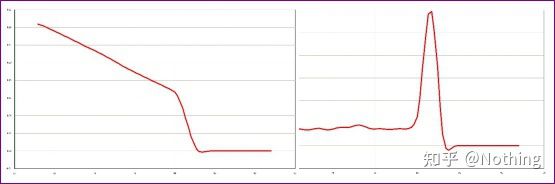
เส้นโค้งการหลอมเหลวปกติ (จุดสูงสุดเดียวที่ไม่มีการขยายแบบไม่เฉพาะเจาะจง)
เส้นโค้งการหลอมละลายที่มีปัญหา (การขยายที่ไม่เฉพาะเจาะจงของยอดเขาปลอม)
【การวิเคราะห์กรณี】
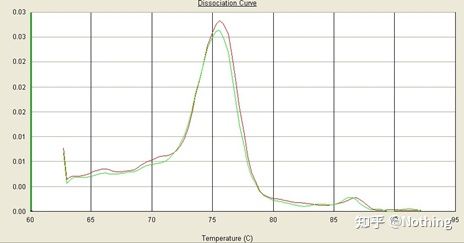
มีพีคหลักแต่ไพรเมอร์หรี่จริงจัง
เส้นโค้งการหลอมละลายจุดสูงสุดเดียวในรูปด้านล่างสามารถหลอกตาของคุณได้ง่ายๆ โดยคิดว่าเป็นการทดลองที่สมบูรณ์แบบ แต่ผลที่ได้นั้นผิดอย่างสิ้นเชิงในเวลานี้เราต้องดูที่อุณหภูมิหลอมเหลวอุณหภูมิสูงสุดต่ำกว่า 80°C ซึ่งเป็นไพรเมอร์-ไดเมอร์โดยสมบูรณ์
ไม่มีชิ้นส่วนเป้าหมาย ไพรเมอร์หรี่ไฟทั้งหมด
นี่พี่หยุดไม่ได้ภาพด้านล่างเป็นภาพที่ถ่ายด้วยโทรศัพท์มือถือที่ส่งมาให้ฉันโดยไอ้พวกขี้โกงน้ำยาที่เขาใช้ล้วนเป็นยี่ห้อที่ใช้กันทั่วไปในอุตสาหกรรมเขาเปลี่ยนจากคำนำหน้าตราตัว T เป็นตราตัว T อีกยี่ห้อหนึ่งฉันคิดว่าคุณเดาได้แล้วคนขี้โกงบ่นกับฉันว่า: "น้ำยาที่ใช้ในภาพแรกนั้นดีเกินไป และจุดพีคก็เป็นเพียงจุดเดียวต่อมาหลังจากใช้รีเอเจนต์ที่คุณแนะนำ มันจะกลายเป็นเหมือนภาพที่สองที่มียอดผสมคุณทำให้ฉันเป็นทุกข์“
แยกกราฟทั้งสองออกจากกันเมื่อมองแวบแรก หนึ่งมีจุดสูงสุดเดียว และอีกจุดหนึ่งมีจุดสูงสุดเป็นสองเท่าเรื่องไร้สาระ แน่นอนว่าจุดสูงสุดเดียวก็ใช้ได้เป็นเรื่องจริงหรือไม่?
แย่กว่า Dou E ถ้าฉันวางสองภาพไว้ในภาพด้านล่างคุณจะเข้าใจทันทีความจริงแล้วภาพแบบนี้ทำให้เราเป็นอัมพาตได้ง่ายหลังจากการวิเคราะห์อย่างระมัดระวัง เราพบว่า: จุดสูงสุดของตัวเลขแรกอยู่ที่ 75°C ซึ่งเป็นไพรเมอร์ไดเมอร์ทั้งหมดจุดสูงสุดของตัวเลขที่สองปรากฏที่ 75°C และ 82°C อย่างน้อยก็มี ผลิตภัณฑ์ปรากฏขึ้น
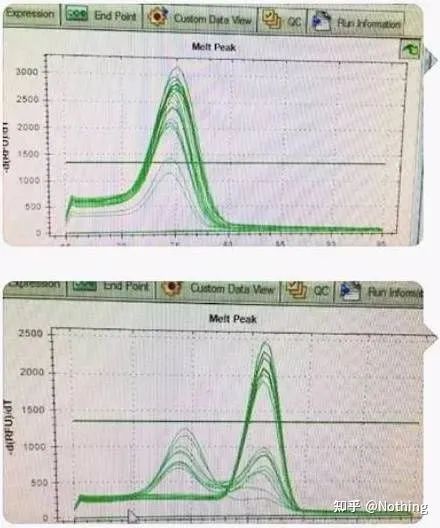
ภาพการตอบรับจากนักศึกษา
ดังนั้นปัญหาพื้นฐานจึงไม่ใช่ปัญหาของรีเอเจนต์ แต่เป็นปัญหาของการออกแบบสีรองพื้นในเวลาเดียวกัน ยังพิสูจน์ได้ว่าแบรนด์ใหญ่ๆ บางแบรนด์ไม่มีคุณภาพที่เป็นเหล็ก และยังพิสูจน์สิ่งที่พี่ชายของฉันพูดก่อนหน้านี้: ไม่ใช่แบรนด์น้ำยาที่ใช้สนับสนุนผลิตภัณฑ์ของคุณเป็นบทความของคุณที่สนับสนุนแบรนด์ของน้ำยาลองนึกดูว่าถ้าพวกขยะไม่เปลี่ยนรีเอเจนต์ ข้อมูลที่ไม่ถูกต้องจะถูกส่งไปยังบันทึกประจำวัน และสิ่งที่จะเกิดขึ้นก็คือโศกนาฏกรรม
2. ค่า Ct ของการควบคุมที่ว่างเปล่า
อย่าอธิบาย ถ้าตัวควบคุมว่างมีค่า Ct มลพิษไม่ใช่หรืออย่างไรก็ตาม คุณยังคงต้องเข้าใจว่าตัวควบคุมว่างใดมีค่า Ctหากเป็นกทช.แสดงว่ามีดีเอ็นเอแปลกปลอม เช่น น้ำยาปนเปื้อนหากเป็น NRT แสดงว่า RNA ที่สกัดได้มี DNA เจือปน
3. เส้นโค้งมาตรฐาน
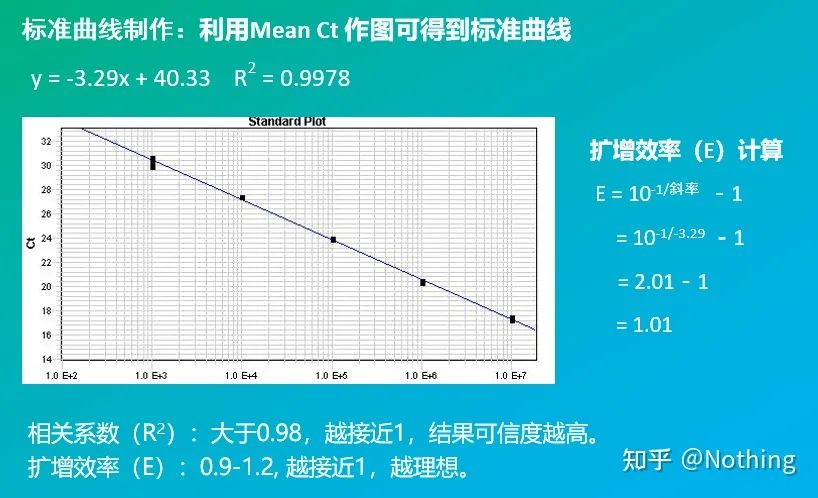
รวมถึงความชันและสูตรการคำนวณ ประสิทธิภาพ PCR สามารถคำนวณผ่านสูตรได้การทดลองที่สมบูรณ์แบบกำหนดให้ความชันของเส้นโค้งมาตรฐานเข้าใกล้ 3.32 และ R² ต้องเข้าใกล้ 0.9999
4. ช่วงไดนามิกเชิงเส้น
ช่วงไดนามิกของปฏิกิริยาเป็นแบบเส้นตรงตามเทมเพลตที่ใช้สร้างเส้นโค้งมาตรฐาน ช่วงไดนามิกควรมีการไล่ระดับความเข้มข้นอย่างน้อย 5 แบบ และให้ความสนใจกับการเปลี่ยนแปลงของค่า Ct ที่การไล่ระดับสีที่มีความเข้มข้นสูงและการไล่ระดับสีที่มีความเข้มข้นต่ำ
5. ความแม่นยำในการตรวจจับ
การเปลี่ยนแปลงในผลลัพธ์ของ qPCR นั่นคือ ความสามารถในการทำซ้ำได้ไม่ดี กล่าวคือ ความแม่นยำไม่ดี มีสาเหตุมาจากหลายปัจจัย รวมถึงอุณหภูมิ ความเข้มข้น และการทำงานความแม่นยำ qPCR โดยทั่วไปจะควบคุมได้น้อยลงเมื่อจำนวนสำเนาลดลงการแปรผันทางเทคนิคนี้ควรจะแตกต่างจากการแปรผันทางชีวภาพ และการจำลองแบบทางชีวภาพสามารถระบุความแตกต่างทางสถิติโดยตรงในผลลัพธ์ qPCR ระหว่างกลุ่มหรือการรักษาโดยเฉพาะอย่างยิ่งสำหรับการตรวจวินิจฉัย จะต้องมีการรายงานความแม่นยำระหว่างการทดสอบ (ความสามารถในการทำซ้ำ) ที่ดีที่สุดทั่วทั้งไซต์และผู้ปฏิบัติงาน
6. ประสิทธิภาพการตรวจจับและ LOD (ในมัลติเพล็กซ์ qPCR)
LOD คือความเข้มข้นต่ำสุดที่ 95% ของตัวอย่างที่ตรวจพบกล่าวอีกนัยหนึ่ง ความเข้มข้นของ LOD ที่อยู่ในชุดของการทำซ้ำของยีนเป้าหมายไม่ควรเกิน 5% ของปฏิกิริยาที่ล้มเหลวเมื่อทำการวิเคราะห์ qPCR แบบมัลติเพล็กซ์ โดยเฉพาะอย่างยิ่งสำหรับการตรวจจับการกลายพันธุ์ของจุดหรือความแตกต่างหลายจุดพร้อมกัน qPCR แบบมัลติเพล็กซ์จำเป็นต้องแสดงหลักฐานว่าความแม่นยำของชิ้นส่วนเป้าหมายหลายชิ้นไม่ได้ลดลงในหลอดเดียวกัน การตรวจจับหลายจุดและการตรวจจับหลอดเดียว ประสิทธิภาพและ LOD ควรเหมือนกันโดยเฉพาะอย่างยิ่งเมื่อมีการขยายยีนเป้าหมายที่มีความเข้มข้นสูงและยีนเป้าหมายที่มีความเข้มข้นต่ำพร้อมกัน ปัญหานี้จะต้องได้รับการเอาใจใส่
ปัญหาและแนวทางแก้ไขโดยทั่วไปแล้ว ปัญหาที่มักพบในการดีบัก qPCR มุ่งเน้นไปที่ประเด็นต่อไปนี้:
·การขยายแบบไม่เฉพาะเจาะจง
· เลือกความเข้มข้นของไพรเมอร์ได้ยากและมีปัญหากับไพรเมอร์-ไดเมอร์
·อุณหภูมิการหลอมไม่ถูกต้อง
· โครงสร้างรองส่งผลต่อประสิทธิภาพการขยายสัญญาณ
การขยายแบบไม่เฉพาะเจาะจง
การขยายที่ไม่เฉพาะเจาะจงเกิดขึ้น โดยทั่วไปถือว่าการออกแบบไพรเมอร์ไม่เหมาะสม แต่ถ้าคุณไม่รีบเปลี่ยนไพรเมอร์ คุณสามารถลองวิธีต่อไปนี้ก่อน (แนบหลักการมาด้วย):
·เพิ่มอุณหภูมิการหลอม – พยายามทำให้ไม่สามารถรักษาพันธะไฮโดรเจนที่อ่อนแอได้
· ลดระยะเวลาการหลอมและการยืดตัว – ลดโอกาสของพันธะไฮโดรเจนที่อ่อนแอ
·ลดความเข้มข้นของไพรเมอร์ – ลดโอกาสในการจับตัวของไพรเมอร์ที่ซ้ำซ้อนและบริเวณที่ไม่ใช่เป้าหมาย
ประสิทธิภาพการขยายต่ำ
สถานการณ์ตรงกันข้ามกับการขยายสัญญาณที่ไม่เฉพาะเจาะจง – ประสิทธิภาพการขยายต่ำ และมาตรการในการจัดการกับการขยายประสิทธิภาพต่ำนั้นตรงกันข้าม:
·ยืดเวลาการหลอมและการยืดตัว
·เปลี่ยนเป็น PCR สามขั้นตอนและลดอุณหภูมิการหลอม
·เพิ่มความเข้มข้นของไพรเมอร์
Ps: นักศึกษาระดับบัณฑิตศึกษาหลายคนที่เกิดในทศวรรษที่ 90 ไม่เต็มใจที่จะศึกษาวิธีการดีบักการทดลอง และหวังว่าชุดทดสอบจะสามารถแก้ปัญหาได้อย่างสมบูรณ์ (ถ้าคุณต้องการไปที่บริษัทน้ำยาทำปฏิกิริยาเพื่อทำการวิจัยและพัฒนาหลังจากสำเร็จการศึกษา) อันที่จริง ผู้ผลิตน้ำยาทำปฏิกิริยาก็คิดแบบนี้เช่นกัน ฉันหวังว่ามันจะโง่เง่า คุณสามารถใช้มันได้ ดังนั้นผู้ผลิตน้ำยาจึงใช้ความพยายามอย่างมากในการแก้ปัญหาการขยายที่ไม่เฉพาะเจาะจง รวมถึงการแนะนำปัจจัยการดูดซับพันธะ H ที่อ่อนแอเพื่อให้แก้ปัญหาได้ง่าย คนเขลายังคงต้องอ่านบทนำของบริษัททำปฏิกิริยาเพื่อดูว่ามีปัจจัยที่ดูดซับพันธะไฮโดรเจนที่อ่อนแอหรือไม่
เลือกความเข้มข้นของไพรเมอร์ได้ยากและมีปัญหากับไพรเมอร์-ไดเมอร์
วิธีที่ 1: โดยทั่วไป ชุดคำสั่งสำหรับ qPCR มีระบบที่แนะนำและความเข้มข้นของไพรเมอร์ที่แนะนำ
วิธีที่ 2: การดีบักโดยการตั้งค่าการไล่ระดับความเข้มข้นของไพรเมอร์รูปภาพด้านล่างถูกขโมยมาจากบริษัทแห่งหนึ่งเพื่ออธิบายรูปด้านล่างแสดงผลลัพธ์เชิงปริมาณของฟลูออเรสเซนซ์ที่ทำด้วยการไล่ระดับความเข้มข้นของไพรเมอร์สามแบบ (100nM, 250nM, 500nM) และการไล่ระดับความเข้มข้นของแม่แบบสี่แบบ (0.1ng, 1ng, 10ng, 100ng)ค่า Ct ของผลการทดลองถูกลงจุดดังนี้:
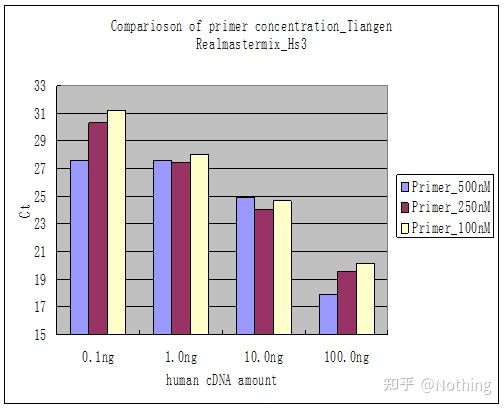
การเลือกความเข้มข้นของสีรองพื้น นำความเข้มข้นของสีรองพื้นแต่ละสีมาเชื่อมกันเป็นเส้นดังนี้:
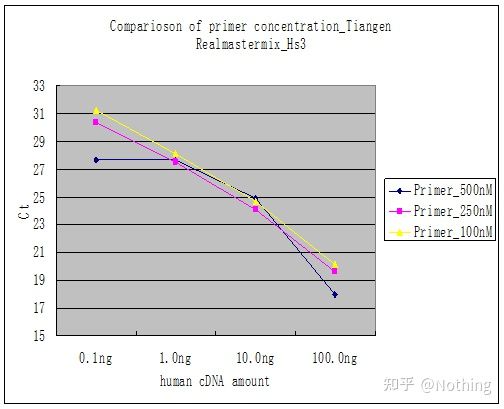
ทางเลือกของความเข้มข้นของไพรเมอร์นั้นชัดเจน ความสัมพันธ์เชิงเส้นของความเข้มข้นของไพรเมอร์ 100nM และ 250nM นั้นดีกว่า และความสัมพันธ์เชิงเส้นของความเข้มข้นของไพรเมอร์ 500nM นั้นค่อนข้างแย่ใน 100nM และ 250nM ค่า Ct ที่ 250nM ค่อนข้างน้อย ดังนั้นความเข้มข้นของไพรเมอร์ที่เหมาะสมคือ 250nMไพรเมอร์-ไดเมอร์ที่รุนแรงโดยทั่วไปสามารถเห็นได้ในเส้นโค้งการหลอมละลายจะเป็นอย่างไรหากไพรเมอร์ที่ออกแบบไม่สามารถหลีกเลี่ยงไพรเมอร์-ไดเมอร์ได้?
วิธีที่ 3: ลดปริมาณไพรเมอร์และเพิ่มอุณหภูมิการหลอม (ไม่ต้องอธิบาย)
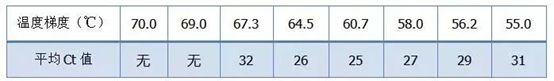
ค่าเชิงประจักษ์ของอุณหภูมิการหลอมคือ 60°Cหากคุณไม่แน่ใจ จะเลือกอุณหภูมิการหลอมที่เหมาะสมได้อย่างไรคำตอบก็เหมือนกับการเลือกความเข้มข้นของไพรเมอร์ –การทดสอบการไล่ระดับสี.ถ่ายภาพจากบริษัท Bio-rad เพื่ออธิบายปัญหาสำหรับการขยายของชิ้นส่วนเป้าหมาย ให้ตั้งค่าการไล่ระดับสีแปดระดับ แต่ละอันมีการทำซ้ำสามครั้ง และเส้นโค้งการขยายที่ได้รับจะเป็นดังนี้:
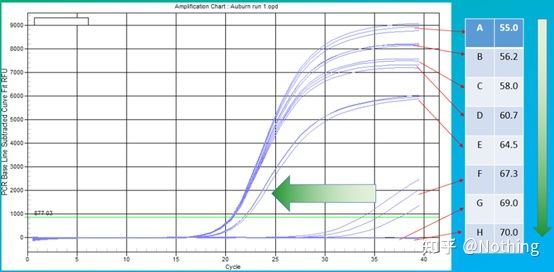
การเลือกอุณหภูมิการหลอม:
·70°C, 69°C—โดยพื้นฐานแล้ว ไพรเมอร์ไม่สามารถรวมกันได้ ดังนั้นจึงไม่มีการขยาย
·67.3°C – มีกำลังขยายเล็กน้อยที่จุดเริ่มต้น และค่า Ct ค่อนข้างใหญ่
·64.5°C——ค่า Ct ลดลง
·ที่ 60.7°C, 58.0°C, 56.2°C และ 55.0°C ค่า Ct มักจะคงที่ แต่ค่าการเรืองแสงสุดท้ายแตกต่างกัน
วิธีการเลือก?หลักการ: หลักการแรกคือค่า Ct ที่สูงขึ้นสำหรับค่า Ct ที่เท่ากัน ให้เลือกอุณหภูมิการหลอมที่สูงขึ้นเพื่อหลีกเลี่ยงการลดขนาดและการขยายที่ไม่เฉพาะเจาะจงแม้ว่าจะมีค่าการเรืองแสงสูงกว่าที่ 55°C แต่อาจมีการหรี่แสงหรือการขยายที่ไม่เฉพาะเจาะจงอยู่ในนั้น
แต่ถ้าคุณฉลาดเท่าคุณ คุณจะคิดว่า: พูดตามเหตุผล ถ้าปฏิกิริยา PCR นั้นเฉพาะเจาะจงมาก ตราบใดที่ความเข้มข้นของไพรเมอร์เกินความต้องการขั้นต่ำ จุดสูงและจุดต่ำสุดก็ไม่ควรมีผล เช่นเดียวกับสีย้อมเรืองแสงและ dNTPอันที่จริง ตราบใดที่อุณหภูมิการหลอมได้รับการปรับปรุงอย่างเหมาะสม ผลกระทบของความเข้มข้นของไพรเมอร์ต่อค่า Ct จะลดลงตามธรรมชาติ
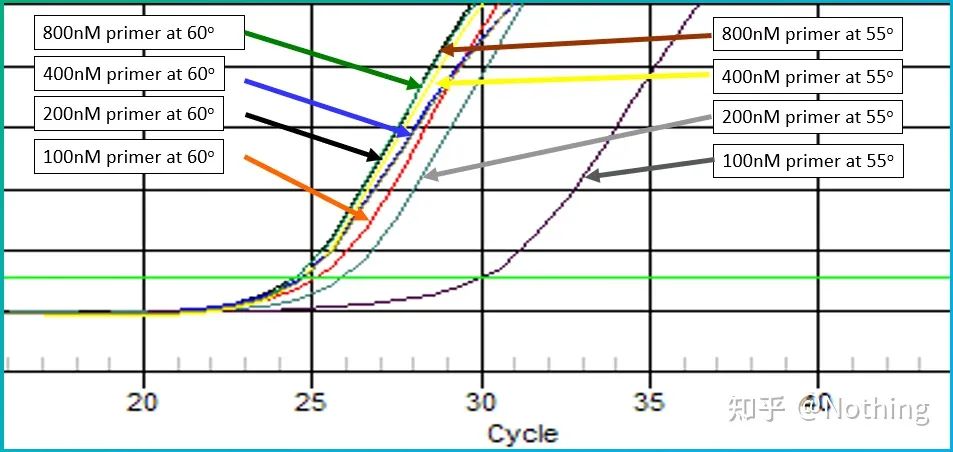
ปรับอุณหภูมิการหลอมอย่างเหมาะสม และผลกระทบของความเข้มข้นของไพรเมอร์ต่อ CT จะลดลง
โครงสร้างทุติยภูมิส่งผลต่อประสิทธิภาพการขยายสัญญาณ
ขอนำภาพจาก Bio-rad มาอธิบายปัญหานอกจากนี้ยังออกแบบการไล่ระดับอุณหภูมิเพื่อขยายยีนที่มีโครงสร้างทุติยภูมิ
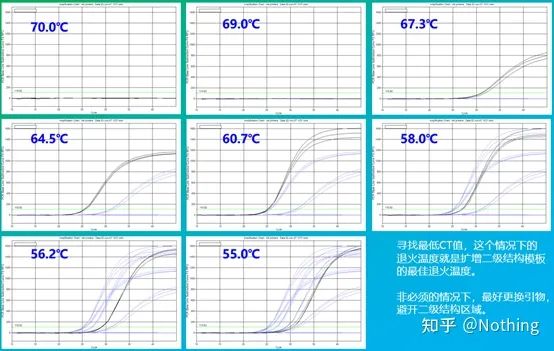
โครงสร้างรองปรากฏขึ้น
จะเห็นได้ว่าเมื่อเกรเดียนต์ของอุณหภูมิลดลง ผลิตภัณฑ์จะเริ่มปรากฏขึ้นและค่า Ct เคลื่อนไปข้างหน้าจนถึงค่าต่ำสุดที่ 60.7°C และเมื่อเกรเดียนต์ของอุณหภูมิลดลง ค่า Ct จะใหญ่ขึ้นในทางกลับกัน เมื่ออุณหภูมิเพิ่มขึ้น โครงสร้างรองจะเปิดขึ้นและประสิทธิภาพการขยายสัญญาณจะเพิ่มขึ้นหลังจากอุณหภูมิถึงระดับหนึ่งแล้ว การเพิ่มอุณหภูมิจะไม่สามารถปรับปรุงประสิทธิภาพการขยายสัญญาณได้เนื่องจากไพรเมอร์ไม่สามารถรวมกันได้อย่างเสถียรในขณะนี้ดังนั้น,มองหาอุณหภูมิที่มีค่า Ct ต่ำสุดซึ่งเป็นอุณหภูมิที่ดีที่สุดสำหรับการขยายแม่แบบโครงสร้างทุติยภูมิ!แน่นอน คนฉลาดโง่ต้องรู้ว่าถ้าไม่จำเป็น ดีที่สุดคือเปลี่ยนไพรเมอร์และหลีกเลี่ยงบริเวณโครงสร้างรอง
5. ระดับแอปพลิเคชัน
MIQE—การวิเคราะห์ข้อมูล

การวิเคราะห์ข้อมูลส่วนใหญ่กำหนดโดยเครื่องมือ PCR เชิงปริมาณเรืองแสงในบทความที่แล้ว มีการวิเคราะห์ข้อมูลจำนวนมาก เช่น ตัวควบคุมเปล่า ซึ่งได้อธิบายไว้ในการออกแบบการทดลองแล้วยีนอ้างอิงภายใน หมายเลขซ้ำ ฯลฯ ได้รับการชี้แจงแล้วในที่นี้เราจะอธิบายถึงการประยุกต์ใช้ qPCR เป็นหลัก
qPCR ถูกนำมาใช้อย่างแพร่หลาย และการทวนสอบเชิงทดลองและการวินิจฉัยกรดนิวคลีอิกเป็นสถานการณ์ที่ใช้บ่อยที่สุด
ปริมาณที่แน่นอน
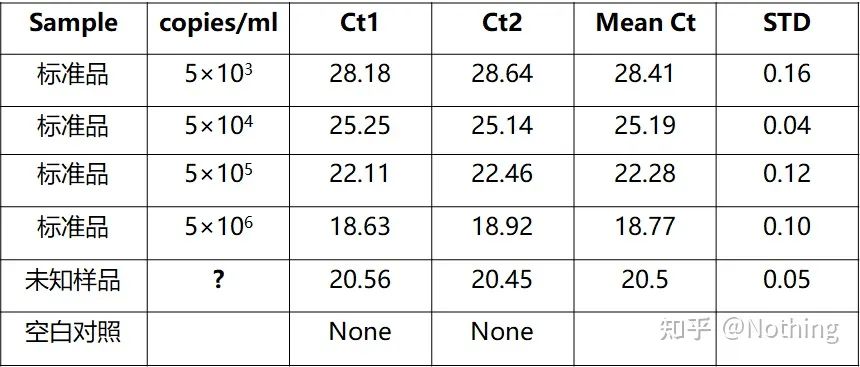
บันทึก (ความเข้มข้นเริ่มต้น) มีความสัมพันธ์เชิงเส้นตรงกับจำนวนรอบสามารถวาดเส้นโค้งมาตรฐานจากมาตรฐานที่ทราบหมายเลขสำเนาเริ่มต้นได้ นั่นคือสามารถหาความสัมพันธ์เชิงเส้นของปฏิกิริยาการขยายสัญญาณได้ตามค่า Ct ของตัวอย่าง สามารถคำนวณความเข้มข้นในตัวอย่างได้จำนวนเทมเพลตที่จะรวม
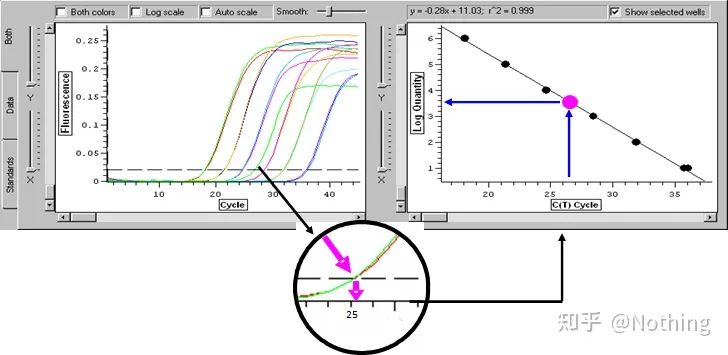
วิธีการคำนวณเชิงปริมาณสัมบูรณ์
การหาปริมาณสัมบูรณ์ต้องขึ้นอยู่กับเส้นโค้งมาตรฐานในการสร้างเส้นโค้งมาตรฐาน จำเป็นต้องมีมาตรฐานโดยปกติแล้วมาตรฐานคือพลาสมิดที่ได้จากการโคลนยีนเป้าหมายทำไมถึงเป็นพลาสมิด?เนื่องจาก DNA พลาสมิดแบบวงกลมนั้นเสถียรที่สุดเจือจางผลิตภัณฑ์มาตรฐานใน 5 ถึง 6 การไล่ระดับสีตามอัตราส่วนสองเท่า (การเจือจาง 10 เท่า) และให้ความสำคัญกับความสม่ำเสมอเมื่อเจือจางให้ค่า Ct อยู่ระหว่าง 15-30
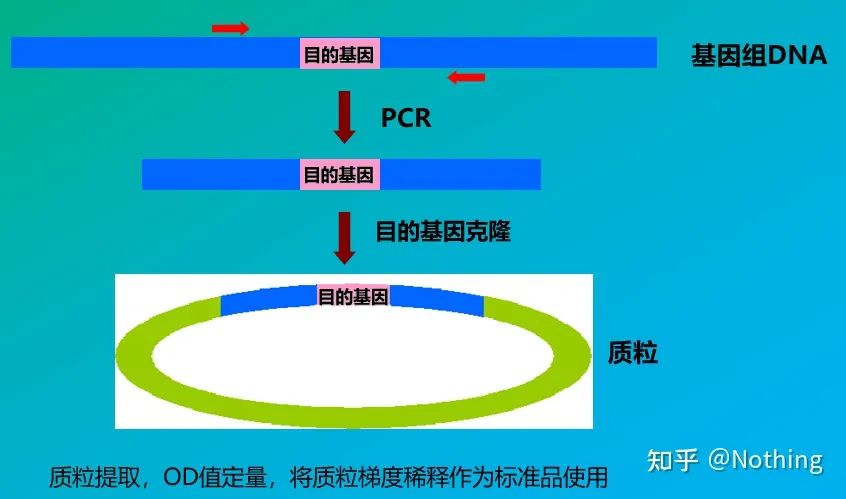
การจัดทำมาตรฐาน
ในเวลาเดียวกัน ตัวอย่างที่จะทดสอบควรเจือจางด้วย (จำปัจจัยการเจือจาง) และค่า Ct ควรอยู่ระหว่าง 15-30นำผลิตภัณฑ์มาตรฐาน + ตัวอย่างที่จะทดสอบมาใส่ในเครื่องพร้อมกันหลังจากการเรียกใช้ เส้นโค้งมาตรฐานถูกสร้างขึ้นด้วยสารมาตรฐาน และตัวอย่างที่จะทดสอบจะถูกนำไปที่เส้นโค้งมาตรฐานเพื่อคำนวณความเข้มข้น
การวัดปริมาณ HBV ของไวรัสตับอักเสบบีเป็นการวัดปริมาณสัมบูรณ์โดยทั่วไป ซึ่งสามารถคำนวณจำนวนสำเนาของไวรัสในเลือด 1 มล.
การคำนวณจำนวนสำเนา
ความเข้มข้นของตัวอย่างที่จะทดสอบ (ng/ul) = OD260 × 50ug/ml × dilution factor
น้ำหนักโมเลกุลของตัวอย่าง = จำนวนเบส × 324
หมายเลขสำเนาของตัวอย่างที่จะทดสอบ (สำเนา/ul) = ความเข้มข้นของตัวอย่างที่จะทดสอบ / น้ำหนักโมเลกุลของตัวอย่าง × 6 × 1014

วิธีการคำนวณจำนวนสำเนา

ข้างต้นเป็นวิธีการคำนวณหาปริมาณนี่เป็นปัญหาทางคณิตศาสตร์ที่สามารถแก้ไขได้หลังจากจบการศึกษาระดับมัธยมต้น และโดยทั่วไปแล้วปัญหาทางคณิตศาสตร์จะแก้ไขได้ด้วยคอมพิวเตอร์ไม่เข้าใจสามารถมาสื่อสารได้
ปริมาณสัมพัทธ์
ปริมาณสัมพัทธ์ส่วนใหญ่จะใช้ในการวิจัยทางวิทยาศาสตร์เลือด 1 มล. มีไวรัสกี่ตัว และเป็นไวรัส DNA นี่เป็นเหตุการณ์ที่ค่อนข้างกำหนดได้: สามารถระบุปริมาณเลือดได้ และไวรัส DNA ค่อนข้างเสถียรอย่างไรก็ตาม เป็นเรื่องยากสำหรับเราที่จะเปรียบเทียบจำนวนสำเนาของการถอดรหัสของยีนบางตัวในลีฟ เนื่องจากเป็นการยากที่จะระบุขนาด น้ำหนัก และความอ่อนโยนของลีฟ ปริมาณของ RNA ที่สกัดออกมานั้นยากที่จะระบุ และประสิทธิภาพของการถอดรหัสแบบย้อนกลับก็ยากที่จะระบุ กล่าวคือ ขั้นตอนใดๆ อาจทำให้ข้อมูลการทดลองมีจุดบกพร่องและไม่สามารถนำมาใช้ได้
ดังนั้น การหาปริมาณสัมพัทธ์จะต้องแนะนำองค์ประกอบ:ยีนอ้างอิงภายใน
กล่าวอีกนัยหนึ่ง ปริมาณสัมพัทธ์คือการเปรียบเทียบระหว่างยีนเป้าหมายกับยีนอ้างอิงภายในเมื่อเปรียบเทียบในเนื้อเยื่อเดียวกันและเซลล์เดียวกัน อิทธิพลของขนาดตัวอย่าง ปริมาณการสกัด RNA ประสิทธิภาพการถอดรหัสย้อนกลับ และประสิทธิภาพของ PCR ค่อนข้างน้อยเนื่องจากขนาดตัวอย่างเล็ก ทั้งยีนอ้างอิงภายในและยีนเป้าหมายจึงค่อนข้างลดลงนี่คือเหตุผลที่เราเน้นความสม่ำเสมอและความมั่นคงมาก่อน
ยีนอ้างอิงภายในโดยทั่วไปยีนแม่บ้านทำความสะอาด(ยีนดูแลบ้าน) ซึ่งหมายถึงกลุ่มของยีนที่แสดงออกอย่างคงที่ในทุกเซลล์ และผลิตภัณฑ์ของยีนเหล่านี้มีความจำเป็นต่อการรักษากิจกรรมชีวิตพื้นฐานของเซลล์
อย่าสับสนกับแนวคิดนี้ยีนการดูแลบ้านเป็นคำศัพท์เกี่ยวกับการทำงานทางชีวภาพ ในขณะที่ยีนอ้างอิงภายในเป็นคำศัพท์ทางเทคนิคเชิงทดลองยีนการดูแลบ้านต้องผ่านการตรวจสอบก่อนจึงจะสามารถเลือกเป็นยีนอ้างอิงภายในได้
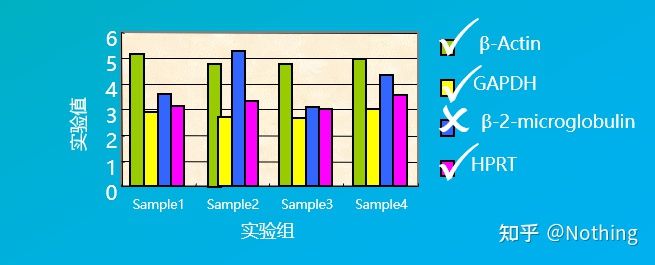
ตัวอย่างเช่น เราเลือกยีนการดูแลบ้านหลายยีนในรูปด้านล่างเพื่อทดสอบระดับการแสดงออกของพวกมันในเซลล์เนื้อเยื่อต่างๆ และพบว่าระดับการแสดงออกของ β-2-ไมโครโกลบูลินค่อนข้างแตกต่างจากยีนของอีกสามยีน ดังนั้นจึงไม่สามารถใช้เป็นยีนอ้างอิงภายในได้
หลังจากทำความเข้าใจเกี่ยวกับฟังก์ชันการแก้ไขของยีนอ้างอิงภายในแล้ว อัลกอริทึมสองรายการจะได้รับมาเนื่องจากการแนะนำของยีนอ้างอิงภายใน
· วิธีโค้งสองมาตรฐาน
·2 – △△Ct method (วิธีการเปรียบเทียบค่า CT)
หากคุณสนใจที่จะศึกษาสปีชีส์และการทำงานของยีน โปรดเลิกการวิจัยเกี่ยวกับอัลกอริธึมและใช้สูตรโดยตรง หรือใช้เครื่องจักรโดยตรงหากคุณเป็นสายตรงในวิชาคณิตศาสตร์และวิศวกรรม โปรดอย่าลังเล
วิธีโค้งสองมาตรฐาน
หาปริมาณยีนเป้าหมายและยีนการดูแลทำความสะอาดของตัวอย่างควบคุมและตัวอย่างที่จะทดสอบผ่านเส้นโค้งมาตรฐาน จากนั้นคำนวณค่าสัมพัทธ์ตามสูตรการคำนวณ ซึ่งเป็นระดับการแสดงออกสัมพัทธ์
ข้อดี: การวิเคราะห์อย่างง่าย การเพิ่มประสิทธิภาพเชิงทดลองที่ค่อนข้างง่าย
ข้อเสีย: สำหรับแต่ละยีน การทดลองแต่ละรอบต้องทำเส้นโค้งมาตรฐาน
การประยุกต์ใช้: หนึ่งในสองวิธีที่ใช้กันมากที่สุดและเป็นที่รู้จักในเชิงปริมาณเชิงปริมาณในการศึกษาการควบคุมการแสดงออกของยีน
สูตรมีดังนี้:

ตัวอย่างมีดังนี้:
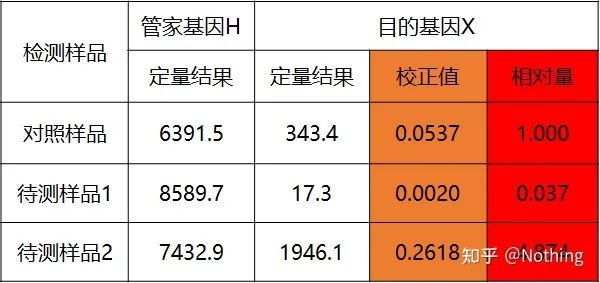
คำนวณจำนวนสัมพัทธ์ตามผลลัพธ์เชิงปริมาณ
2 – วิธี△△Ct (วิธีการเปรียบเทียบค่า CT)

ข้อดี: ไม่ต้องสร้างเส้นโค้งมาตรฐาน
ข้อเสีย: สันนิษฐานว่าประสิทธิภาพการขยายใกล้ถึง 100%;ส่วนเบี่ยงเบนมาตรฐานคือ < 5% และเส้นโค้งมาตรฐานและประสิทธิภาพระหว่างการขยายแต่ละครั้งจะถือว่าสอดคล้องกันการปรับเงื่อนไขการทดลองให้เหมาะสมนั้นซับซ้อนกว่า
การประยุกต์ใช้: หนึ่งในสองวิธีที่ใช้กันมากที่สุดและเป็นที่รู้จักในเชิงปริมาณเชิงปริมาณในการศึกษาการควบคุมการแสดงออกของยีน
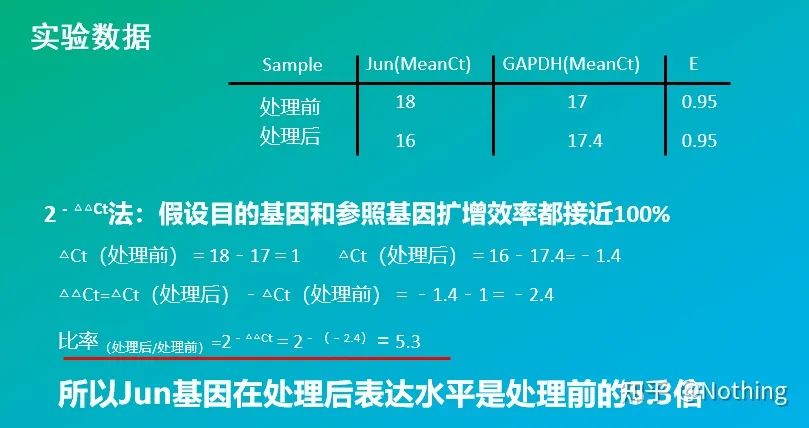
แน่นอน ประสิทธิภาพการขยายมักจะเป็นไปไม่ได้อย่างสมบูรณ์ 1. วิธีแก้ไข: หากเรารู้ว่ายีนเป้าหมายและยีนอ้างอิงมีประสิทธิภาพการขยายเท่ากัน แต่ประสิทธิภาพการขยายไม่เท่ากับ 1 ดังนั้น 2-△△Ct สามารถแก้ไขเป็น: (1+E )-△△Ct ตัวอย่างเช่น หากประสิทธิภาพการขยายเป็น 0.95 ดังนั้นสูตรการคำนวณสามารถแก้ไขเป็น 1.9 5-△△กะรัต
จนถึงตอนนี้ เนื้อหาเกี่ยวกับ PCR เชิงปริมาณเรืองแสงได้สิ้นสุดลงแล้ว
เวลาโพสต์: เม.ย.-06-2023



