



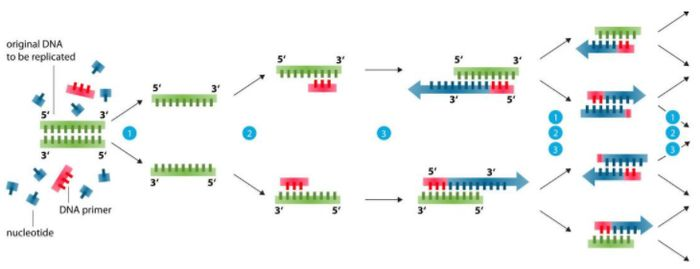
พื้นฐานการออกแบบรองพื้น (แก้ปัญหาได้ 99%)
1. ความยาวรองพื้น: หนังสือเรียนต้องการ 15-30bp ปกติประมาณ 20bpสภาวะที่แท้จริงควรอยู่ที่ 18-24bp เพื่อให้แน่ใจถึงความจำเพาะ แต่ยิ่งนานยิ่งดี ยิ่งไพรเมอร์ที่ยาวเกินไปจะลดความจำเพาะ และทำให้ผลผลิตลดลงด้วย
2. ช่วงขยายของ Primer: 200-500bp เหมาะสม และสามารถขยายส่วนย่อยได้ถึง 10kb ภายใต้เงื่อนไขเฉพาะ
3. ฐานรองพื้น: เนื้อหาของ G+C ควรเป็น 40-60% ผลการขยาย G+C น้อยเกินไปไม่ดี G+C มากเกินไปจะปรากฏแถบที่ไม่เฉพาะเจาะจงได้ง่ายATGC กระจายอย่างสุ่มได้ดีที่สุด หลีกเลี่ยงการรวมกลุ่มของนิวคลีโอไทด์ purine หรือ pyrimidine มากกว่า 5 ตัวMulti-gc สำหรับปลาย 5′ และลำดับกลางเพื่อเพิ่มความเสถียร หลีกเลี่ยง GC ที่สมบูรณ์ที่ปลาย 3′ ไม่มี GC สำหรับ 3 ฐานสุดท้าย หรือไม่มี GC สำหรับ 3 ใน 5 ฐานสุดท้าย
4. หลีกเลี่ยงโครงสร้างรองในไพรเมอร์ และหลีกเลี่ยงการเสริมระหว่างไพรเมอร์สองตัว โดยเฉพาะการเสริมที่ปลาย 3 ' มิฉะนั้นจะเกิดไพรเมอร์หรี่และแถบขยายที่ไม่เฉพาะเจาะจงจะถูกสร้างขึ้น
5. ฐานที่ปลาย 3 ' ของไพรเมอร์ โดยเฉพาะฐานสุดท้ายและฐานรองสุดท้าย ควรจับคู่อย่างเคร่งครัดเพื่อหลีกเลี่ยงความล้มเหลวของ PCR เนื่องจากฐานของขั้วต่อที่ไม่ได้จับคู่
6. ไพรเมอร์มีหรือสามารถเพิ่มด้วยตำแหน่งการตัดแยกที่เหมาะสม และลำดับเป้าหมายที่ขยายอย่างพึงประสงค์ควรมีตำแหน่งการตัดแยกที่เหมาะสม ซึ่งเป็นประโยชน์อย่างมากสำหรับการวิเคราะห์การตัดแยกหรือการโคลนโมเลกุล
7. ความจำเพาะของไพรเมอร์: ไพรเมอร์ไม่ควรมีความคล้ายคลึงกันอย่างชัดเจนกับลำดับอื่นๆ ในฐานข้อมูลลำดับกรดนิวคลีอิก
8. เรียนรู้การใช้ซอฟต์แวร์: PP5, Oligo6, DNAstar, Vector NTI, primer3 (การออกแบบออนไลน์นี้ทำงานได้ดีที่สุด)
เนื้อหาข้างต้นสามารถแก้ปัญหาการออกแบบสีรองพื้นได้อย่างน้อย 99%
ควบคุมรายละเอียดการออกแบบสีรองพื้น
1. ความยาวของไพรเมอร์
ความยาวของไพรเมอร์ทั่วไปคือ 18~30 ฐานโดยทั่วไป ปัจจัยที่สำคัญที่สุดที่กำหนดอุณหภูมิการหลอมของไพรเมอร์คือความยาวของไพรเมอร์โดยทั่วไปจะเลือกอุณหภูมิการหลอมของไพรเมอร์ (ค่า Tm -5℃) และบางส่วนใช้ค่า Tm โดยตรงสามารถใช้สูตรต่อไปนี้เพื่อคำนวณอุณหภูมิการหลอมของไพรเมอร์อย่างคร่าวๆ
เมื่อความยาวของไพรเมอร์น้อยกว่า 20bp: [4(G+C)+2(A+T)]-5℃
เมื่อความยาวของสีรองพื้นมากกว่า 20bp: 62.3℃+0.41℃(%GC)-500/ความยาว-5℃
นอกจากนี้ยังสามารถใช้ซอฟต์แวร์จำนวนมากในการคำนวณอุณหภูมิการหลอม หลักการคำนวณจะแตกต่างกัน ดังนั้นบางครั้งค่าที่คำนวณได้อาจมีช่องว่างเล็กน้อยเพื่อเพิ่มประสิทธิภาพปฏิกิริยา PCR ไพรเมอร์ที่สั้นที่สุดซึ่งรับประกันอุณหภูมิการหลอมไม่ต่ำกว่า 54 ℃ ถูกนำมาใช้เพื่อประสิทธิภาพและความจำเพาะสูงสุด
โดยรวมแล้ว ความจำเพาะของไพรเมอร์เพิ่มขึ้นสี่เท่าสำหรับแต่ละนิวคลีโอไทด์เพิ่มเติม ดังนั้นความยาวไพรเมอร์ขั้นต่ำสำหรับการใช้งานส่วนใหญ่คือ 18 นิวคลีโอไทด์ขีดจำกัดบนของความยาวไพรเมอร์นั้นไม่สำคัญมากนัก ส่วนใหญ่เกี่ยวข้องกับประสิทธิภาพของปฏิกิริยาเนื่องจากเอนโทรปี ยิ่งไพรเมอร์ยาวเท่าไร อัตราการหลอมตัวเพื่อจับกับ DNA เป้าหมายก็จะยิ่งต่ำลงเพื่อสร้างแม่แบบเกลียวคู่ที่เสถียรสำหรับ DNA polymerase ในการจับกัน
เมื่อใช้ซอฟต์แวร์ในการออกแบบไพรเมอร์ ความยาวของไพรเมอร์สามารถกำหนดได้ด้วยค่า TM โดยเฉพาะอย่างยิ่งสำหรับไพรเมอร์ของ PCR เชิงปริมาณเรืองแสง ควรควบคุม TM=60℃ หรือมากกว่านั้น
เนื้อหา 2.GC
โดยทั่วไป เนื้อหาของ G+C ในลำดับไพรเมอร์คือ 40%~60% และควรประสานเนื้อหา GC และค่า Tm ของไพรเมอร์คู่หนึ่งหากไพรเมอร์มีแนวโน้ม GC หรือ AT ที่รุนแรง ให้เพิ่มหาง A, T หรือ G และ C ในปริมาณที่เหมาะสมที่ปลาย 5 ' ของไพรเมอร์
3. อุณหภูมิการหลอม
อุณหภูมิการหลอมควรต่ำกว่าอุณหภูมิคลายโซ่ 5 ℃หากจำนวนของฐานไพรเมอร์มีน้อย อุณหภูมิการหลอมก็สามารถเพิ่มได้อย่างเหมาะสม ซึ่งจะเพิ่มความจำเพาะของ PCR ได้หากจำนวนฐานมากก็สามารถลดอุณหภูมิการหลอมได้อย่างเหมาะสมความแตกต่างของอุณหภูมิการหลอมระหว่างไพรเมอร์ 4 ℃ ~ 6 ℃ จะไม่ส่งผลต่อผลผลิต PCR แต่ตามหลักแล้วอุณหภูมิการหลอมของไพรเมอร์ 1 คู่จะเท่ากัน ซึ่งอาจแตกต่างกันระหว่าง 55 ℃ ~ 75 ℃
4. หลีกเลี่ยงพื้นที่โครงสร้างรองของเทมเพลตการขยายเสียง
ทางที่ดีควรหลีกเลี่ยงบริเวณโครงสร้างรองของเทมเพลตเมื่อเลือกส่วนขยายโครงสร้างรองที่เสถียรของแฟรกเมนต์เป้าหมายสามารถคาดการณ์และประเมินได้โดยซอฟต์แวร์คอมพิวเตอร์ที่เกี่ยวข้อง ซึ่งจะเป็นประโยชน์สำหรับการเลือกเทมเพลตผลการทดลองแสดงให้เห็นว่าการขยายมักไม่ประสบผลสำเร็จเมื่อพลังงานอิสระ (△G) ของบริเวณที่จะขยายมีค่าน้อยกว่า 58.6lkJ/mol
5. ไม่ตรงกับ DNA เป้าหมาย
เมื่อลำดับ DNA เป้าหมายที่ถูกขยายมีขนาดใหญ่ ไพรเมอร์อาจจับกับหลายส่วนของ DNA เป้าหมาย ส่งผลให้เกิดแถบหลายแถบปรากฏขึ้นในผลลัพธ์เวลานี้จำเป็นต้องใช้การทดสอบซอฟต์แวร์ BLAST เว็บไซต์:http://www.ncbi.nlm.nih.gov/BLAST/.เลือกจัดแนวสองลำดับ (bl2seq)
การวางลำดับไพรเมอร์ไปยังโซน 1 และลำดับดีเอ็นเอเป้าหมายไปยังโซน 2 นั้นสามารถใช้แทนกันได้ และ BLAST จะคำนวณคอมพลีเมนต์ แอนติเซนส์ และความเป็นไปได้อื่นๆ ดังนั้นผู้ใช้จึงไม่จำเป็นต้องสังเกตว่าโซ่ทั้งสองเป็นเซนส์เชนหรือไม่คุณยังสามารถป้อนหมายเลข GI หากคุณทราบหมายเลข GI ของลำดับในฐานข้อมูล คุณจึงไม่ต้องวางลำดับส่วนใหญ่สุดท้าย คลิกจัดตำแหน่งที่ 3 เพื่อดูว่าไพรเมอร์มีไซต์ที่คล้ายคลึงกันหลายไซต์ใน DNA เป้าหมายหรือไม่
6. ขั้วไพรเมอร์
ปลาย 3 'ของไพรเมอร์คือจุดเริ่มต้นของส่วนขยาย ดังนั้นจึงเป็นเรื่องสำคัญที่จะต้องป้องกันไม่ให้จุดเริ่มต้นที่ไม่ตรงกันปลาย 3 'ไม่ควรเกิน 3 G หรือ C ติดต่อกัน เนื่องจากจะทำให้ไพรเมอร์ทำงานผิดพลาดในภูมิภาคลำดับการเพิ่มคุณค่า G+Cปลาย 3 'ไม่สามารถสร้างโครงสร้างทุติยภูมิได้ ยกเว้นในปฏิกิริยา PCR พิเศษ (AS-PCR) ปลาย 3' ของไพรเมอร์ไม่สามารถจับคู่กันได้ตัวอย่างเช่น หากมีการขยายขอบเขตการเข้ารหัส ปลาย 3 ' ของไพรเมอร์ไม่ควรสิ้นสุดที่ตำแหน่งที่สามของ codon เนื่องจากตำแหน่งที่สามของ codon มีแนวโน้มที่จะเสื่อมสภาพ ซึ่งจะส่งผลต่อความจำเพาะและประสิทธิภาพของการขยายสัญญาณเมื่อใช้ไพรเมอร์เสริม โปรดดูตารางการใช้โคดอน ให้ความสนใจกับการตั้งค่าทางชีวภาพ อย่าใช้ไพรเมอร์เสริมที่ปลาย 3′ และใช้ไพรเมอร์ที่มีความเข้มข้นสูงกว่า (1uM-3uM)
7. โครงสร้างรองของไพรเมอร์
ไพรเมอร์เองไม่ควรมีลำดับเสริม มิฉะนั้น ไพรเมอร์เองจะพับเป็นโครงสร้างกิ๊บ และโครงสร้างรองนี้จะส่งผลต่อการยึดเกาะของไพรเมอร์และแม่แบบเนื่องจากสิ่งกีดขวางแบบสเตอริกหากใช้การตัดสินแบบประดิษฐ์ เบสเสริมที่ต่อเนื่องของไพรเมอร์เองไม่ควรเกิน 3bpไม่ควรมีการเสริมกันระหว่างไพรเมอร์ทั้งสองโดยเฉพาะอย่างยิ่งควรหลีกเลี่ยงการทับซ้อนกันของปลาย 3 'เพื่อป้องกันการก่อตัวของไพรเมอร์ไดเมอร์โดยทั่วไป ไม่ควรมีเบสที่เหมือนกันหรือคอมพลีเมนต์ริตี้ติดต่อกันเกิน 4 เบสระหว่างไพรเมอร์ 1 คู่
8. เพิ่มเครื่องหมายหรือตำแหน่ง
ปลาย 5 ' มีผลเพียงเล็กน้อยต่อความจำเพาะของการขยาย ดังนั้นสามารถแก้ไขได้โดยไม่กระทบต่อความจำเพาะของการขยายการปรับเปลี่ยนไพรเมอร์ 5 'end รวม: การเพิ่มไซต์ข้อ จำกัด ของเอนไซม์;ไบโอตินที่ติดฉลาก, ฟลูออเรสเซนซ์, ดิจอกซิน, Eu3+ ฯลฯ แนะนำลำดับดีเอ็นเอที่จับกับโปรตีนแนะนำตำแหน่งการกลายพันธุ์ การแทรกและขาดหายไปของลำดับการกลายพันธุ์ และการแนะนำลำดับโปรโมเตอร์ เป็นต้น เบสที่เกินมาจะส่งผลต่อประสิทธิภาพของการขยายสัญญาณไม่มากก็น้อย และเพิ่มโอกาสของการก่อตัวของไพรเมอร์ไดเมอร์ แต่ต้องมีการผ่อนปรนสำหรับขั้นตอนต่อไปลำดับเพิ่มเติมที่ไม่มีอยู่ในลำดับเป้าหมาย เช่น ไซต์ข้อจำกัดและลำดับโปรโมเตอร์ สามารถเพิ่มที่ปลาย 5′ ของไพรเมอร์ได้โดยไม่ส่งผลต่อความเฉพาะเจาะจงลำดับเหล่านี้ไม่รวมอยู่ในการคำนวณค่า Tm ของไพรเมอร์ แต่ควรทดสอบเพื่อหาส่วนประกอบเสริมและโครงสร้างทุติยภูมิภายใน
9. โคลนย่อย
ส่วนใหญ่แล้ว PCR เป็นเพียงการโคลนเบื้องต้นเท่านั้น จากนั้นเราจำเป็นต้องโคลนย่อยชิ้นส่วนเป้าหมายเป็นเวกเตอร์ต่างๆ เราจึงจำเป็นต้องออกแบบฐานเพิ่มเติมสำหรับการดำเนินการถัดไปในขั้นตอน PCR
ลำดับบางส่วนที่ออกแบบมาสำหรับการโคลนย่อยสรุปไว้ด้านล่าง
เพิ่มไซต์ข้อจำกัดของเอ็นโดนิวคลีเอสที่จำกัดแล้ว
การเพิ่มไซต์จำกัดเอนไซม์เป็นวิธีที่ใช้บ่อยที่สุดสำหรับผลิตภัณฑ์ PCR แบบย่อยโดยทั่วไปไซต์ความแตกแยกมีหกฐานนอกเหนือไปจากจุดสิ้นสุดของไซต์ความแตกแยก 5 'จำเป็นต้องเพิ่มฐานป้องกัน 2 ~ 3อย่างไรก็ตาม จำนวนของฐานป้องกันที่เอนไซม์ต่างๆ ต้องการนั้นแตกต่างกันตัวอย่างเช่น SalⅠ ไม่ต้องการฐานป้องกัน, EcoRⅤ ต้องการฐานป้องกัน 1 แห่ง, NotⅠ ต้องการฐานป้องกัน 2 แห่ง และ Hind Ⅲ ต้องการฐานป้องกัน 3 แห่ง
LIC เพิ่มส่วนท้าย
ชื่อเต็มของ LIC คือ Ligation-Independent cloning ซึ่งเป็นวิธีการโคลนที่คิดค้นโดย Navogen โดยเฉพาะสำหรับส่วนของเวกเตอร์ petตัวพา PET ที่เตรียมโดยวิธี LIC มีปลายเหนียวเส้นเดี่ยว 12-15 ฐานที่ไม่เสริม ซึ่งเสริมปลายเหนียวที่สอดคล้องกันบนชิ้นส่วนเม็ดมีดเป้าหมายเพื่อวัตถุประสงค์ในการขยาย ลำดับไพรเมอร์ 5 ′ของชิ้นส่วนที่แทรกควรเสริมเวกเตอร์ LICกิจกรรมการแยกส่วน 3′→5′ ของ T4 DNA polymerase สามารถสร้างปลายเหนียวเส้นเดียวบนชิ้นส่วนที่แทรกเข้าไปหลังจากเวลาอันสั้นเนื่องจากผลิตภัณฑ์สามารถขึ้นรูปได้จากการหลอมร่วมกันของชิ้นส่วนเม็ดมีดและเวคเตอร์ที่เตรียมไว้ วิธีการนี้จึงรวดเร็วและมีประสิทธิภาพมาก และเป็นการโคลนนิ่งโดยตรง
กำกับ TA โคลนเพิ่มหาง
การโคลน TA ไม่สามารถกำหนดเป้าหมายชิ้นส่วนเป็นเวกเตอร์ได้ ดังนั้นในภายหลัง Invitrogen จึงแนะนำเวกเตอร์ที่สามารถกำหนดเป้าหมายไปที่การโคลน ซึ่งประกอบด้วย GTGGS พื้นฐานสี่ตัวที่ปลายด้านหนึ่งดังนั้น ในการออกแบบไพรเมอร์ PCR จึงควรเพิ่มลำดับเสริมตามนั้น เพื่อให้ชิ้นส่วนสามารถ "จัดตำแหน่ง" ได้
หากคุณมีเวลาน้อย คุณสามารถลองสังเคราะห์โดยตรง โดยรวมยีนเข้ากับเวกเตอร์ ซึ่งเรียกว่าการสังเคราะห์ยีน ET ในนักกล้ามเนื้อ
D. วิธีการโคลนฟิวชัน
ไม่ต้องใช้ลิกาส ไม่ต้องมีปฏิกิริยานานตราบเท่าที่มีการใช้ลำดับที่ปลายทั้งสองของเวกเตอร์เชิงเส้นในการออกแบบไพรเมอร์ ผลิตภัณฑ์ PCR และเวกเตอร์เชิงเส้นจะถูกเพิ่มลงในสารละลายเอนไซม์ฟิวชันที่มี BSA และวางไว้ที่อุณหภูมิห้องเป็นเวลาครึ่งชั่วโมง การเปลี่ยนแปลงสามารถทำได้วิธีนี้เหมาะอย่างยิ่งสำหรับการแปลงปริมาณมาก
10. ผสานไพรเมอร์
บางครั้ง ข้อมูลลำดับที่จำกัดเท่านั้นที่ทราบเกี่ยวกับการออกแบบไพรเมอร์ตัวอย่างเช่น หากทราบเฉพาะลำดับกรดอะมิโน ก็สามารถออกแบบไพรเมอร์ที่ผสานได้ไพรเมอร์การควบรวมเป็นส่วนผสมของลำดับที่แตกต่างกันซึ่งแสดงถึงความเป็นไปได้ของเบสที่แตกต่างกันทั้งหมดที่เข้ารหัสกรดอะมิโนตัวเดียวหากต้องการเพิ่มความเฉพาะเจาะจง คุณสามารถดูตารางการใช้โคดอนเพื่อลดการผนวกตามการตั้งค่าการใช้งานพื้นฐานของสิ่งมีชีวิตต่างๆHypoxanthine สามารถจับคู่กับเบสทั้งหมดเพื่อลดอุณหภูมิการหลอมของไพรเมอร์อย่าใช้ฐานที่ต่อท้ายที่ปลาย 3 'ของไพรเมอร์เนื่องจากการหลอมฐาน 3 ฐานสุดท้ายที่ปลาย 3' นั้นเพียงพอที่จะเริ่ม PCR ที่ไซต์ที่ไม่ถูกต้องมีการใช้ไพรเมอร์ที่มีความเข้มข้นสูงกว่า (1μM ถึง 3μM) เนื่องจากไพรเมอร์ในส่วนผสมที่ผนวกเข้าด้วยกันจำนวนมากไม่เฉพาะเจาะจงกับเทมเพลตเป้าหมาย
วัตถุดิบพีซีอาร์ควบคุม
1. ปริมาณรองพื้น
ความเข้มข้นของไพรเมอร์แต่ละตัวคือ 0.1 ~ 1umol หรือ 10 ~ 100pmolเป็นการดีกว่าที่จะสร้างผลลัพธ์ที่ต้องการโดยใช้ไพรเมอร์ในปริมาณที่น้อยที่สุดไพรเมอร์ที่มีความเข้มข้นสูงจะทำให้การขยายไม่ตรงกันและไม่เฉพาะเจาะจง และเพิ่มโอกาสในการเกิดไดเมอร์ระหว่างไพรเมอร์
2. ความเข้มข้นของไพรเมอร์
ความเข้มข้นของไพรเมอร์มีผลต่อความจำเพาะความเข้มข้นของไพรเมอร์ที่เหมาะสมโดยทั่วไปอยู่ระหว่าง 0.1 ถึง 0.5μMความเข้มข้นของไพรเมอร์ที่สูงขึ้นนำไปสู่การขยายผลิตภัณฑ์ที่ไม่เฉพาะเจาะจง
3. อุณหภูมิการหลอมของไพรเมอร์
ตัวแปรที่สำคัญอีกประการหนึ่งสำหรับไพรเมอร์คืออุณหภูมิหลอมเหลว (Tm)นี่คืออุณหภูมิเมื่อ 50% ของไพรเมอร์และลำดับคู่สมแสดงเป็นโมเลกุลดีเอ็นเอสายคู่จำเป็นต้องใช้ Tm ในการตั้งค่าอุณหภูมิการหลอม PCRตามหลักการแล้ว อุณหภูมิการหลอมจะต่ำเพียงพอเพื่อให้แน่ใจว่าการหลอมไพรเมอร์อย่างมีประสิทธิภาพด้วยลำดับเป้าหมาย แต่สูงพอที่จะลดการจับตัวที่ไม่เฉพาะเจาะจงอุณหภูมิการหลอมที่เหมาะสมจาก 55 ℃ถึง 70 ℃โดยทั่วไปอุณหภูมิการหลอมจะตั้งไว้ต่ำกว่า Tm ของไพรเมอร์ 5 ℃
มีหลายสูตรสำหรับการตั้งค่า Tm ซึ่งแตกต่างกันมากขึ้นอยู่กับสูตรที่ใช้และลำดับของไพรเมอร์เนื่องจากสูตรส่วนใหญ่ให้ค่า Tm โดยประมาณ อุณหภูมิการหลอมทั้งหมดจึงเป็นเพียงจุดเริ่มต้นเท่านั้นสามารถปรับปรุงความจำเพาะได้โดยการวิเคราะห์ปฏิกิริยาต่างๆ ที่ทำให้อุณหภูมิการหลอมเพิ่มขึ้นอย่างต่อเนื่องเริ่มต้นที่ต่ำกว่า Tm-5℃ โดยประมาณ และค่อยๆ เพิ่มอุณหภูมิการหลอมทีละ 2℃อุณหภูมิการหลอมที่สูงขึ้นจะลดการก่อตัวของไพรเมอร์ไดเมอร์และผลิตภัณฑ์ที่ไม่เฉพาะเจาะจงเพื่อผลลัพธ์ที่ดีที่สุด ไพรเมอร์ทั้งสองควรมีค่า Tm โดยประมาณหากความแตกต่างของ Tm ของคู่ไพรเมอร์มากกว่า 5℃ ไพรเมอร์จะแสดงการเริ่มต้นที่ผิดพลาดอย่างมีนัยสำคัญโดยใช้อุณหภูมิการหลอมที่ต่ำกว่าในวงจรหากไพรเมอร์สองตัว Tm ต่างกัน ให้ตั้งอุณหภูมิการหลอมให้ต่ำกว่าค่า Tm ต่ำสุด 5 ℃อีกทางหนึ่ง เพื่อเพิ่มความจำเพาะ สามารถทำได้ห้ารอบก่อนที่อุณหภูมิการหลอมออกแบบสำหรับ Tm ที่สูงกว่า ตามด้วยรอบที่เหลือที่อุณหภูมิการหลอมที่ออกแบบมาสำหรับ Tm ที่ต่ำกว่าสิ่งนี้ทำให้สามารถรับสำเนาบางส่วนของเทมเพลตปลายทางได้ภายใต้เงื่อนไขที่เข้มงวด
4. ความบริสุทธิ์และความคงตัวของไพรเมอร์
ความบริสุทธิ์มาตรฐานของไพรเมอร์แบบกำหนดเองนั้นเพียงพอสำหรับการใช้งาน PCR ส่วนใหญ่การกำจัดหมู่เบนโซอิลและไอโซบิวทิลโดยการแยกเกลือออกนั้นน้อยมาก ดังนั้นจึงไม่รบกวน PCRแอปพลิเคชันบางอย่างต้องการการทำให้บริสุทธิ์เพื่อลบลำดับที่ไม่เต็มความยาวในกระบวนการสังเคราะห์ลำดับที่ถูกตัดทอนเหล่านี้เกิดขึ้นเนื่องจากประสิทธิภาพของการสังเคราะห์ DNA ทางเคมีไม่เต็ม 100%นี่เป็นกระบวนการแบบวงกลมที่ใช้ปฏิกิริยาเคมีซ้ำๆ เมื่อแต่ละเบสถูกเติมเพื่อสร้าง DNA จาก 3 ′ถึง 5′คุณสามารถล้มเหลวในทั้งสองรอบไพรเมอร์ที่ยาวขึ้น โดยเฉพาะเบสที่มากกว่า 50 เบส มีสัดส่วนของลำดับที่ถูกตัดออกมาก และอาจต้องมีการทำให้บริสุทธิ์
ผลผลิตของไพรเมอร์ได้รับผลกระทบจากประสิทธิภาพของเคมีสังเคราะห์และวิธีการทำให้บริสุทธิ์บริษัทชีวเวชภัณฑ์ เช่น Cytology และ Shengong ต่างใช้หน่วย OD ขั้นต่ำเพื่อให้แน่ใจว่าผลผลิตทั้งหมดของโอลิโกนิวคลีโอไซด์ไพรเมอร์แบบกำหนดเองจัดส่งในรูปแบบผงแห้งเป็นการดีที่สุดที่จะละลายไพรเมอร์อีกครั้งใน TE เพื่อให้ความเข้มข้นสุดท้ายคือ 100μMTE ดีกว่าน้ำปราศจากไอออน เนื่องจากค่า pH ของน้ำมักจะเป็นกรด และจะทำให้เกิดการไฮโดรไลซิสของโอลิโกนิวคลีโอไซด์
ความเสถียรของสีรองพื้นขึ้นอยู่กับสภาวะการเก็บรักษาควรเก็บผงแห้งและไพรเมอร์ที่ละลายน้ำไว้ที่อุณหภูมิ -20 ℃ไพรเมอร์ที่ละลายใน TE ที่ความเข้มข้นมากกว่า 10μM สามารถเก็บไว้ได้อย่างเสถียรที่ -20°C เป็นเวลา 6 เดือน แต่สามารถเก็บไว้ที่อุณหภูมิห้อง (15°C ถึง 30°C) ได้น้อยกว่า 1 สัปดาห์ไพรเมอร์แบบผงแห้งสามารถเก็บไว้ที่อุณหภูมิ -20 C เป็นเวลาอย่างน้อย 1 ปี และที่อุณหภูมิห้อง (15 C ถึง 30 C) ได้นานถึง 2 เดือน
5. เอนไซม์และความเข้มข้นของเอนไซม์
ในปัจจุบัน Taq DNA polymerase ที่ใช้โดยพื้นฐานแล้วเป็นเอนไซม์วิศวกรรมยีนที่สังเคราะห์โดยแบคทีเรียโคลิฟอร์มปริมาณของเอนไซม์ที่จำเป็นในการเร่งปฏิกิริยา PCR โดยทั่วไปคือประมาณ 2.5U (หมายถึงปริมาตรปฏิกิริยาทั้งหมด 100ul)หากความเข้มข้นสูงเกินไป อาจนำไปสู่การขยายที่ไม่เฉพาะเจาะจงหากความเข้มข้นต่ำเกินไปจะทำให้ปริมาณผลิตภัณฑ์สังเคราะห์ลดลง
6. คุณภาพและความเข้มข้นของ dNTP
คุณภาพของ dNTP นั้นสัมพันธ์อย่างใกล้ชิดกับความเข้มข้นและประสิทธิภาพของการขยาย PCRผง dNTP เป็นเม็ดเล็ก ๆ และความแปรปรวนของมันจะสูญเสียฤทธิ์ทางชีวภาพหากเก็บไว้อย่างไม่เหมาะสมสารละลาย dNTP เป็นกรด และควรใช้ในความเข้มข้นสูง โดยใช้สารละลายบัฟเฟอร์ NaOH 1M หรือ Tris.HCL 1M เพื่อปรับค่า pH เป็น 7.0 ~ 7.5 บรรจุภัณฑ์ย่อยจำนวนน้อย การเก็บรักษาแบบแช่แข็งที่อุณหภูมิ -20℃การละลายน้ำแข็งหลายครั้งจะทำให้ dNTP ลดลงในปฏิกิริยา PCR dNTP ควรอยู่ที่ 50 ~ 200umol/Lโดยเฉพาะอย่างยิ่งควรให้ความสนใจกับความเข้มข้นของ DNTPS ทั้งสี่ควรเท่ากัน (การเตรียมโมลเท่ากัน)หากความเข้มข้นของสารตัวใดตัวหนึ่งแตกต่างจากตัวอื่น (สูงหรือต่ำกว่า) จะทำให้เกิดความไม่ตรงกันความเข้มข้นต่ำเกินไปจะทำให้ผลผลิตของผลิตภัณฑ์ PCR ลดลงdNTP สามารถรวมกับ Mg2+ และลดความเข้มข้นของ Mg2+ อิสระ
7. เทมเพลต (ยีนเป้าหมาย) กรดนิวคลีอิก
ปริมาณและระดับการทำให้บริสุทธิ์ของกรดนิวคลีอิกแม่แบบเป็นหนึ่งในจุดเชื่อมโยงสำคัญสำหรับความสำเร็จหรือความล้มเหลวของ PCRวิธีการทำให้บริสุทธิ์ของ DNA แบบดั้งเดิมมักใช้ SDS และโปรตีเอส K ในการย่อยและกำจัดตัวอย่างหน้าที่หลักของ SDS คือ: ละลายลิพิดและโปรตีนบนเยื่อหุ้มเซลล์ ดังนั้นทำลายเยื่อหุ้มเซลล์โดยการละลายโปรตีนของเยื่อหุ้มเซลล์ และแยกนิวเคลียสโปรตีนในเซลล์ออก SDS ยังสามารถรวมตัวกับโปรตีนและตกตะกอนProtease K สามารถไฮโดรไลซ์และย่อยโปรตีน โดยเฉพาะฮิสโตนที่จับกับ DNA จากนั้นใช้ตัวทำละลายอินทรีย์ฟีนอลและคลอโรฟอร์มเพื่อสกัดโปรตีนและส่วนประกอบอื่นๆ ของเซลล์ และใช้เอทานอลหรือไอโซโพรพิลแอลกอฮอล์เพื่อตกตะกอนกรดนิวคลีอิกกรดนิวคลีอิกที่สกัดออกมาสามารถใช้เป็นแม่แบบสำหรับปฏิกิริยา PCRสำหรับตัวอย่างการตรวจทางคลินิกทั่วไป วิธีการที่ง่ายและรวดเร็วสามารถใช้ในการละลายเซลล์ ไลเซทเชื้อโรค ย่อยและกำจัดโปรตีนออกจากโครโมโซมเพื่อปลดปล่อยยีนเป้าหมาย และใช้โดยตรงสำหรับการขยาย PCRการแยกเทมเพลต RNA มักจะใช้วิธี guanidine isothiocyanate หรือ protease K เพื่อป้องกันไม่ให้ RNase ย่อยสลาย RNA
ความเข้มข้น 8.Mg2+
Mg2+ มีผลอย่างมากต่อความจำเพาะและผลผลิตของการขยาย PCRในปฏิกิริยา PCR ทั่วไป เมื่อความเข้มข้นของ dNTP ต่างๆ คือ 200umol/L ความเข้มข้นที่เหมาะสมของ Mg2+ คือ 1.5 ~ 2.0mmol/Lความเข้มข้นของ Mg2+ สูงเกินไป ความจำเพาะของปฏิกิริยาลดลง การขยายตัวที่ไม่จำเพาะเกิดขึ้น ความเข้มข้นที่ต่ำเกินไปจะลดการทำงานของ Taq DNA polymerase ส่งผลให้ผลิตภัณฑ์จากปฏิกิริยาลดลง
ไอออนของแมกนีเซียมส่งผลต่อ PCR หลายด้าน เช่น กิจกรรมของ DNA polymerase ซึ่งส่งผลต่อผลผลิตอีกตัวอย่างหนึ่งคือการหลอมสีรองพื้นซึ่งส่งผลต่อความจำเพาะdNTP และเทมเพลตจับกับแมกนีเซียมไอออน ลดปริมาณแมกนีเซียมไอออนอิสระที่จำเป็นสำหรับการทำงานของเอนไซม์ความเข้มข้นของแมกนีเซียมไอออนที่เหมาะสมจะแตกต่างกันไปตามคู่ไพรเมอร์และเทมเพลตที่แตกต่างกัน แต่ความเข้มข้นเริ่มต้นของ PCR ทั่วไปที่ 200μM dNTP คือ 1.5mM (หมายเหตุ: สำหรับ PCR เชิงปริมาณแบบเรียลไทม์ ให้ใช้สารละลายแมกนีเซียมไอออน 3 ถึง 5mM พร้อมโพรบเรืองแสง)ความเข้มข้นที่สูงขึ้นของไอออนแมกนีเซียมอิสระจะเพิ่มผลผลิต แต่ยังเพิ่มการขยายแบบไม่เฉพาะเจาะจงและลดความเที่ยงตรงเพื่อกำหนดความเข้มข้นที่เหมาะสมที่สุด การไทเทรตแมกนีเซียมไอออนถูกดำเนินการโดยเพิ่มทีละ 0.5 มิลลิโมลาร์จาก 1 มิลลิโมลาร์ถึง 3 มิลลิโมลาร์เพื่อลดการพึ่งพาการเพิ่มประสิทธิภาพแมกนีเซียมไอออน สามารถใช้ Platinum Taq DNA polymerase ได้Platinum Taq DNA polymerase สามารถรักษาการทำงานในช่วงความเข้มข้นของแมกนีเซียมไอออนที่กว้างกว่า Taq DNA polymerase ดังนั้นจึงต้องการการปรับให้เหมาะสมน้อยกว่า
9. สารเติมแต่งส่งเสริม Pcr
การปรับอุณหภูมิการหลอม การออกแบบไพรเมอร์ และความเข้มข้นของแมกนีเซียมไอออนให้เหมาะสมนั้นเพียงพอสำหรับการขยายความเฉพาะเจาะจงสูงของแม่แบบส่วนใหญ่อย่างไรก็ตาม เทมเพลตบางตัว รวมถึงเทมเพลตที่มีเนื้อหา GC สูง จำเป็นต้องมีมาตรการเพิ่มเติมสารเติมแต่งที่ส่งผลต่ออุณหภูมิหลอมเหลวของ DNA เป็นอีกวิธีหนึ่งในการปรับปรุงความจำเพาะและผลผลิตของผลิตภัณฑ์จำเป็นต้องมีการทำให้แม่แบบเสียหายทั้งหมดเพื่อให้ได้ผลลัพธ์ที่ดีที่สุด
นอกจากนี้ โครงสร้างทุติยภูมิยังช่วยป้องกันการจับตัวของไพรเมอร์และการขยายตัวของเอนไซม์
สารเติมแต่ง PCR รวมถึง formamide, DMSO, กลีเซอรีน, เบทาอีน และ PCRx Enhancer Solution ช่วยเพิ่มการขยายสัญญาณกลไกที่เป็นไปได้ของพวกมันคือการลดอุณหภูมิหลอมเหลว จึงช่วยการหลอมไพรเมอร์และช่วยขยาย DNA polymerase ผ่านบริเวณโครงสร้างทุติยภูมิPCRx Solution มีข้อได้เปรียบอื่นๆจำเป็นต้องมีการเพิ่มประสิทธิภาพแมกนีเซียมไอออนให้น้อยที่สุดเมื่อใช้กับ Platinum Taq DNA polymerase และ Platinum Pfx DNA polymeraseดังนั้น เทคนิคแพลทินัมจึงถูกรวมเข้ากับสารเติมแต่งเพื่อเพิ่มความจำเพาะในขณะที่ลดการพึ่งพาแนวทางที่สาม การปรับแมกนีเซียมไอออนให้เหมาะสมเพื่อให้ได้ผลลัพธ์ที่ดีที่สุด ควรปรับความเข้มข้นของสารเติมแต่งให้เหมาะสม โดยเฉพาะ DMSO, formamide และ glycerol ซึ่งยับยั้ง Taq DNA polymerase

10. เริ่มร้อนแรง
Hot start PCR เป็นหนึ่งในวิธีการที่สำคัญที่สุดในการปรับปรุงความจำเพาะของ PCR นอกเหนือไปจากการออกแบบไพรเมอร์ที่ดีแม้ว่าอุณหภูมิการยืดตัวที่เหมาะสมที่สุดของ Taq DNA polymerase คือ 72 ℃ แต่โพลิเมอเรสยังคงทำงานอยู่ที่อุณหภูมิห้องดังนั้น ผลิตภัณฑ์ที่ไม่เฉพาะเจาะจงจะถูกผลิตขึ้นเมื่ออุณหภูมิในการจับยึดต่ำกว่าอุณหภูมิการหลอมในระหว่างการเตรียมปฏิกิริยา PCR และที่จุดเริ่มต้นของวัฏจักรความร้อนผลิตภัณฑ์ที่ไม่เฉพาะเจาะจงเหล่านี้จะถูกขยายอย่างมีประสิทธิภาพHot-start PCR มีประสิทธิภาพโดยเฉพาะอย่างยิ่งเมื่อตำแหน่งที่ใช้สำหรับการออกแบบไพรเมอร์ถูกจำกัดโดยตำแหน่งขององค์ประกอบทางพันธุกรรม เช่น การกลายพันธุ์ที่ควบคุมตำแหน่ง การโคลนนิพจน์ หรือการสร้างและการจัดการองค์ประกอบทางพันธุกรรมที่ใช้สำหรับวิศวกรรมดีเอ็นเอ
วิธีทั่วไปในการจำกัดกิจกรรมของ Taq DNA polymerase คือการเตรียมสารละลายปฏิกิริยา PCR บนน้ำแข็งและวางลงในเครื่อง PCR ที่อุ่นไว้ล่วงหน้าวิธีนี้ง่ายและราคาไม่แพง แต่ไม่ทำให้การทำงานของเอนไซม์สมบูรณ์ ดังนั้นจึงไม่สามารถกำจัดการขยายตัวของผลิตภัณฑ์ที่ไม่เฉพาะเจาะจงได้อย่างสมบูรณ์
ไพรมิ่งด้วยความร้อนจะชะลอการสังเคราะห์ DNA โดยยับยั้งส่วนประกอบที่สำคัญจนกว่าเครื่องมือ PCR จะถึงอุณหภูมิที่เสียสภาพธรรมชาติวิธีการเริ่มต้นความร้อนด้วยตนเองส่วนใหญ่ รวมถึงการเติม Taq DNA polymerase ที่ล่าช้านั้นยุ่งยาก โดยเฉพาะอย่างยิ่งสำหรับการใช้งานปริมาณงานสูงวิธีการรองพื้นด้วยความร้อนอื่นๆ ใช้แว็กซ์ชีลด์เพื่อปิดล้อมส่วนประกอบที่สำคัญ รวมถึงแมกนีเซียมไอออนหรือเอนไซม์ หรือเพื่อแยกส่วนประกอบที่เกิดปฏิกิริยาทางกายภาพ เช่น แม่แบบและบัฟเฟอร์ในระหว่างรอบการระบายความร้อน ส่วนประกอบต่างๆ จะถูกปล่อยออกมาและผสมเข้าด้วยกันเมื่อขี้ผึ้งละลายเช่นเดียวกับวิธีการเริ่มร้อนด้วยตนเอง วิธีแว็กซ์ชิลด์นั้นยุ่งยากและมีแนวโน้มที่จะเกิดการปนเปื้อน และไม่เหมาะสำหรับการใช้งานปริมาณงานสูง
Platinum DNA polymerase สะดวกและมีประสิทธิภาพสำหรับ PCR แบบเริ่มร้อนอัตโนมัติPlatinum Taq DNA polymerase ประกอบด้วย Taq DNA polymerase รีคอมบิแนนท์รวมกับโมโนโคลนอลแอนติบอดีต่อ Taq DNA polymeraseแอนติบอดีถูกสร้างสูตรโดย PCR เพื่อยับยั้งการทำงานของเอนไซม์ระหว่างการคงอุณหภูมิเป็นเวลานานTaq DNA polymerase ถูกปล่อยออกมาในปฏิกิริยาระหว่างฉนวน 94 ℃ของขั้นตอนการเสียสภาพธรรมชาติ คืนค่ากิจกรรมของพอลิเมอเรสทั้งหมดตรงกันข้ามกับ Taq DNA polymerase ที่ดัดแปลงทางเคมีสำหรับการเริ่มต้นความร้อน เอนไซม์แพลทินัมไม่ต้องการฉนวนเป็นเวลานานที่ 94 ℃ (10 ถึง 15 นาที) เพื่อกระตุ้นโพลีเมอเรสด้วย PlatinumTaq DNA polymerase 90% ของกิจกรรม Taq DNA polymerase ได้รับการกู้คืนหลังจาก 2 นาทีที่ 94 ℃

11. รัง-PCR
การขยายรอบต่อเนื่องโดยใช้ไพรเมอร์ที่ซ้อนกันสามารถปรับปรุงความจำเพาะและความไวรอบแรกเป็นการขยายมาตรฐาน 15 ถึง 20 รอบเศษเล็กเศษน้อยของผลิตภัณฑ์ขยายเริ่มต้นถูกเจือจาง 100 ถึง 1,000 เท่า และเพิ่มรอบที่สองของการขยายเป็น 15 ถึง 20 รอบอีกทางหนึ่งคือ ผลิตภัณฑ์ขยายขนาดเริ่มต้นสามารถปรับขนาดได้โดยการทำให้บริสุทธิ์ด้วยเจลไพรเมอร์แบบซ้อนจะใช้ในการขยายรอบที่สอง ซึ่งสามารถผูกกับลำดับเป้าหมายภายในไพรเมอร์ตัวแรกการใช้ PCR ที่ซ้อนกันช่วยลดความเป็นไปได้ในการขยายไซต์เป้าหมายหลายแห่ง เนื่องจากมีลำดับเป้าหมายไม่กี่ชุดที่เสริมกับไพรเมอร์ทั้งสองชุดจำนวนรอบทั้งหมดเท่ากัน (30 ถึง 40) ด้วยไพรเมอร์เดียวกันขยายไซต์ที่ไม่เฉพาะเจาะจงNested PCR จะเพิ่มความไวของลำดับเป้าหมายที่จำกัด (เช่น mrnas ที่หายาก) และปรับปรุงความจำเพาะของ PCRS ที่ยาก (เช่น 5 ′RACE)
12. PCR จากมากไปน้อย
PCR จากมากไปน้อยช่วยปรับปรุงความจำเพาะโดยใช้สภาวะการหลอมที่แน่นสำหรับ PCR สองสามรอบแรกวัฏจักรเริ่มต้นที่อุณหภูมิการหลอมสูงกว่าค่า Tm ประมาณ 5 ℃ จากนั้นแต่ละรอบจะลดลง 1 ℃ถึง 2 ℃ จนกว่าอุณหภูมิการหลอมจะต่ำกว่า Tm 5 ℃เฉพาะเทมเพลตปลายทางที่มีความคล้ายคลึงกันสูงสุดเท่านั้นที่จะถูกขยายผลิตภัณฑ์เหล่านี้ยังคงขยายตัวต่อไปในรอบต่อ ๆ ไป ทำให้ผลิตภัณฑ์ที่ไม่เฉพาะเจาะจงขยายตัวออกไปPCR จากมากไปน้อยมีประโยชน์สำหรับวิธีการที่ไม่ทราบระดับความคล้ายคลึงกันระหว่างไพรเมอร์และแม่แบบเป้าหมาย เช่น ลายนิ้วมือ AFLP DNA
ชุด PCR ที่เกี่ยวข้อง
ฮีโร่ 2× PCRTMระบบผสมมีความทนทานต่อสารยับยั้ง PCR สูงกว่าระบบผสม PCR ทั่วไป และสามารถรับมือกับการขยาย PCR ของเทมเพลตที่ซับซ้อนต่างๆ ได้อย่างง่ายดายระบบปฏิกิริยาที่ไม่เหมือนใครและ Taq Hero ที่มีประสิทธิภาพสูงทำให้ปฏิกิริยา PCR มีประสิทธิภาพการขยายความเฉพาะเจาะจงและความไวที่สูงขึ้น
ประสิทธิภาพการขยายที่สูงขึ้น
มีกิจกรรม DNA polymerase 5'→3' และกิจกรรม exonuclease 5'→3' โดยไม่มีกิจกรรม exonuclease 3'→5'
PCR แบบเรียลไทม์ Easyᵀᴹ-SYBR Green I Kit
เฉพาะ - บัฟเฟอร์ที่ปรับให้เหมาะสมและเอนไซม์ Taq ที่เริ่มร้อนสามารถป้องกันการขยายที่ไม่เฉพาะเจาะจงและการก่อตัวของไพรเมอร์ไดเมอร์
ความไวสูง—สามารถตรวจหาแม่แบบที่มีสำเนาต่ำได้
RT-PCR Easyᵀᴹ I (ขั้นตอนเดียว)
ชุดเครื่องมือนี้ใช้รีเอเจนต์การถอดรหัสแบบย้อนกลับของ Foregene และ Foregene HotStar Taq DNA Polymerase รวมกับระบบปฏิกิริยาที่ไม่เหมือนใครเพื่อปรับปรุงประสิทธิภาพการขยายและความจำเพาะของปฏิกิริยาได้อย่างมีประสิทธิภาพ
เวลาโพสต์: May-09-2023



