



RT-qPCR ได้รับการพัฒนาจากเทคโนโลยี PCR ธรรมดาเพิ่มสารเคมีเรืองแสง (สีย้อมเรืองแสงหรือโพรบเรืองแสง) ให้กับระบบปฏิกิริยา PCR แบบดั้งเดิม และตรวจจับกระบวนการหลอมและขยาย PCR ตามเวลาจริงตามกลไกการเรืองแสงที่แตกต่างกันการเปลี่ยนแปลงสัญญาณเรืองแสงในตัวกลางใช้ในการคำนวณปริมาณการเปลี่ยนแปลงของผลิตภัณฑ์ในแต่ละรอบของ PCRปัจจุบัน วิธีการทั่วไปคือวิธีย้อมเรืองแสงและวิธีโพรบ
วิธีการย้อมเรืองแสง:
สีย้อมเรืองแสงบางชนิด เช่น SYBR Green Ⅰ, PicoGreen, BEBO เป็นต้น ไม่เปล่งแสงด้วยตัวมันเอง แต่จะเปล่งแสงหลังจากจับกับร่องเล็ก ๆ ของ dsDNAดังนั้นในช่วงเริ่มต้นของปฏิกิริยา PCR เครื่องจึงไม่สามารถตรวจจับสัญญาณเรืองแสงได้เมื่อปฏิกิริยาดำเนินต่อไปจนถึงการขยายการหลอม (วิธีสองขั้นตอน) หรือขั้นตอนการขยาย (วิธีสามขั้นตอน) สายคู่จะเปิดขึ้นในเวลานี้ และ DNA พอลิเมอเรสใหม่ในระหว่างการสังเคราะห์เส้นใย โมเลกุลเรืองแสงจะรวมกันในร่องย่อย dsDNA และเปล่งแสงเรืองแสงเมื่อจำนวนของรอบ PCR เพิ่มขึ้น สีย้อมจะรวมกับ dsDNA มากขึ้นเรื่อยๆ และสัญญาณเรืองแสงก็ได้รับการปรับปรุงอย่างต่อเนื่องเช่นกันใช้ SYBR Green Ⅰ เป็นตัวอย่าง
วิธีการสอบสวน:
โพรบ Taqman เป็นโพรบไฮโดรไลซิสที่ใช้บ่อยที่สุดมีกลุ่มฟลูออเรสเซนต์ที่ปลายโพรบ 5 นิ้ว ซึ่งโดยปกติจะเป็น FAMโพรบนั้นเป็นลำดับที่เสริมกับยีนเป้าหมายมีกลุ่มดับเรืองแสงที่ปลาย 3 ′ของฟลูออโรฟอร์ตามหลักการของการถ่ายโอนพลังงานเรโซแนนซ์เรืองแสง (Försterเรโซแนนซ์การถ่ายโอนพลังงาน FRET) เมื่อกลุ่มเรืองแสงนักข่าว (โมเลกุลเรืองแสงของผู้บริจาค) และกลุ่มเรืองแสงดับ (โมเลกุลเรืองแสงตัวรับ) เมื่อสเปกตรัมกระตุ้นทับซ้อนกันและระยะทางอยู่ใกล้มาก (7-10 นาโนเมตร) การกระตุ้นของโมเลกุลผู้บริจาคสามารถกระตุ้นการเรืองแสงของโมเลกุลตัวรับ ในขณะที่การเรืองแสงอัตโนมัตินั้น อ่อนแอลงดังนั้นที่จุดเริ่มต้นของปฏิกิริยา PCR เมื่อโพรบว่างและไม่เสียหายในระบบ กลุ่มสารเรืองแสงนักข่าวจะไม่ปล่อยสารเรืองแสงเมื่อทำการหลอม ไพรเมอร์และโพรบจะเชื่อมกับแม่แบบในระหว่างขั้นตอนการขยาย พอลิเมอเรสจะสังเคราะห์สายโซ่ใหม่อย่างต่อเนื่องDNA polymerase มีกิจกรรม exonuclease 5′-3′เมื่อไปถึงโพรบ DNA พอลิเมอเรสจะไฮโดรไลซ์โพรบจากแม่แบบ แยกกลุ่มเรืองแสงรีพอร์ตออกจากกลุ่มเควนเชอร์ฟลูออเรสเซนต์ แล้วปล่อยสัญญาณเรืองแสงเนื่องจากโพรบและแม่แบบมีความสัมพันธ์แบบหนึ่งต่อหนึ่ง วิธีการโพรบจึงดีกว่าวิธีย้อมในแง่ของความแม่นยำและความไวของการทดสอบ
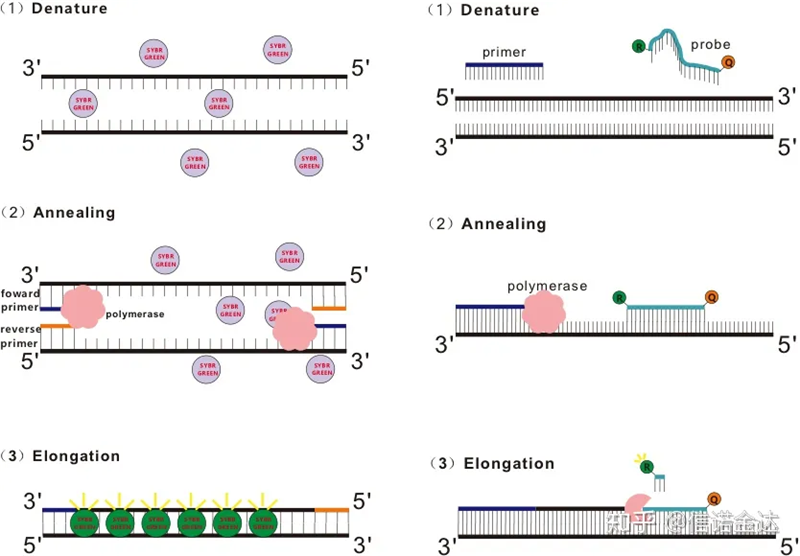
รูปที่ 1 หลักการของ qRT-PCR
การออกแบบรองพื้น
หลักการ:
ไพรเมอร์ควรได้รับการออกแบบในบริเวณที่สงวนไว้ของชุดกรดนิวคลีอิกและมีความจำเพาะ
วิธีที่ดีที่สุดคือใช้ลำดับ cDNA และลำดับ mRNA ก็เป็นที่ยอมรับเช่นกันหากไม่มี ให้ค้นหาการออกแบบบริเวณซีดีของลำดับดีเอ็นเอ
ความยาวของผลิตภัณฑ์เชิงปริมาณเรืองแสงคือ 80-150bp ยาวที่สุดคือ 300bp ความยาวไพรเมอร์โดยทั่วไปอยู่ระหว่าง 17-25 เบส และความแตกต่างระหว่างไพรเมอร์ต้นน้ำและปลายน้ำไม่ควรใหญ่เกินไป
เนื้อหา G+C อยู่ระหว่าง 40% ถึง 60% และ 45-55% ดีที่สุด
ค่า TM อยู่ระหว่าง 58-62 องศา
พยายามหลีกเลี่ยงไพรเมอร์ไดเมอร์และเซลฟ์ไดเมอร์ (อย่าให้เบสเสริมติดต่อกันเกิน 4 คู่) หากหลีกเลี่ยงไม่ได้ ให้สร้าง ΔG<4.5kJ/mol* หากคุณไม่สามารถแน่ใจว่าได้ลบ gDNA ระหว่างการถอดความแบบย้อนกลับ สะอาด วิธีที่ดีที่สุดคือการออกแบบไพรเมอร์ของปลาย intron *3′ ไม่สามารถแก้ไขได้ และเพื่อหลีกเลี่ยง AT, GC บริเวณที่อุดม ให้หลีกเลี่ยง T/C, A/G โครงสร้างต่อเนื่อง (2-3) และไพรเมอร์ที่ไม่ใช่ -
เฉพาะเจาะจง โฮโมโลยีของลำดับที่ขยายต่างกันอย่างพึงประสงค์คือน้อยกว่า 70% หรือมี 8 โฮโมโลยีเบสประกอบ
ฐานข้อมูล:
ค้นหา CottonFGD ด้วยคำหลัก
การออกแบบรองพื้น:
การออกแบบไพรเมอร์ IDT-qPCR
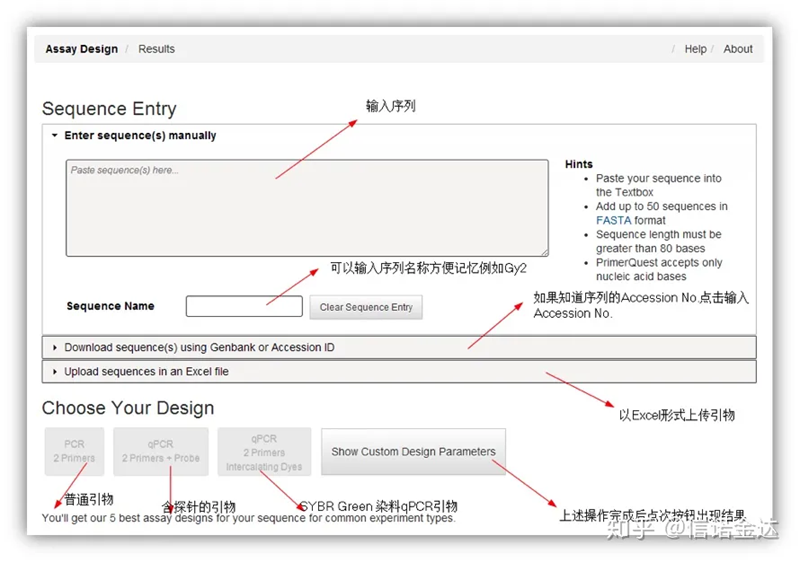
รูปที่ 2 หน้าเครื่องมือออกแบบไพรเมอร์ออนไลน์ IDT
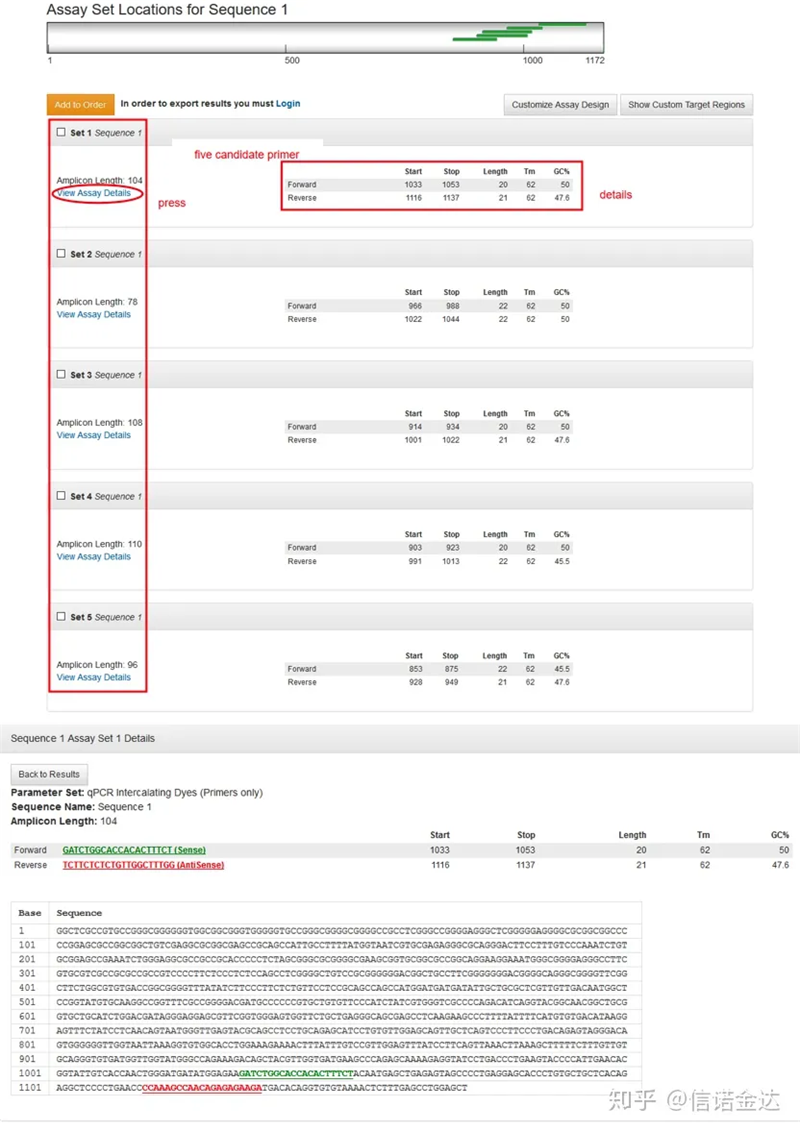
การแสดงผลหน้าผลลัพธ์รูปที่ 3
การออกแบบไพรเมอร์ lncRNA:
lncRNA:ขั้นตอนเดียวกับ mRNA
miRNA:หลักการของวิธีสเต็มลูป: เนื่องจาก miRNA ทั้งหมดเป็นลำดับสั้นๆ ประมาณ 23 nt จึงไม่สามารถดำเนินการตรวจหา PCR โดยตรงได้ ดังนั้นจึงใช้เครื่องมือลำดับสเต็มลูปลำดับสเต็มลูปคือ DNA สายเดี่ยวประมาณ 50 nt ซึ่งสามารถสร้างโครงสร้างกิ๊บได้เอง3 'ส่วนท้ายสามารถออกแบบเป็นลำดับที่เสริมกับชิ้นส่วนย่อยของ miRNA จากนั้นสามารถเชื่อมต่อ miRNA เป้าหมายกับลำดับสเต็มลูประหว่างการถอดความแบบย้อนกลับ และความยาวทั้งหมดสามารถเข้าถึง 70bp ซึ่งสอดคล้องกับความยาวของผลิตภัณฑ์ขยายที่กำหนดโดย qPCRการออกแบบไพรเมอร์สำหรับหางแร่ miRNA
การตรวจจับเฉพาะการขยาย:
ฐานข้อมูลการระเบิดออนไลน์: การระเบิดของ CottonFGD ตามลำดับความคล้ายคลึงกัน
Local blast: อ้างถึงการใช้ Blast+ เพื่อทำการ blast ในเครื่อง, linux และ macos สามารถสร้างฐานข้อมูลในเครื่องได้โดยตรง, ระบบ win10 ยังสามารถทำได้หลังจากติดตั้ง ubuntu bashสร้างฐานข้อมูลการระเบิดในพื้นที่และการระเบิดในพื้นที่เปิด ubuntu bash บน win10
ข้อสังเกต: ฝ้ายที่ดอนและฝ้ายที่เกาะทะเลเป็นพืช tetraploid ดังนั้นผลลัพธ์ของการระเบิดมักจะตรงกันสองอย่างขึ้นไปในอดีต การใช้ NAU cds เป็นฐานข้อมูลเพื่อทำการ blast มีแนวโน้มที่จะพบยีนที่คล้ายคลึงกัน 2 ยีนที่มีความแตกต่างของ SNP เพียงเล็กน้อยโดยปกติแล้ว ยีนที่คล้ายคลึงกันทั้งสองไม่สามารถแยกออกได้ด้วยการออกแบบไพรเมอร์ ดังนั้นจึงถือว่าพวกมันเหมือนกันหากมีอินเดลที่ชัดเจน ไพรเมอร์มักจะออกแบบบนอินเดล แต่สิ่งนี้อาจนำไปสู่โครงสร้างรองของไพรเมอร์ พลังงานอิสระจะสูงขึ้น ส่งผลให้ประสิทธิภาพการขยายลดลง แต่ก็หลีกเลี่ยงไม่ได้
การตรวจจับโครงสร้างทุติยภูมิของไพรเมอร์:
ขั้นตอน:เปิด oligo 7 → ป้อนลำดับเทมเพลต → ปิดหน้าต่างย่อย → บันทึก → ค้นหาตำแหน่งไพรเมอร์บนเทมเพลต กด ctrl+D เพื่อกำหนดความยาวของไพรเมอร์ → วิเคราะห์โครงสร้างทุติยภูมิต่างๆ เช่น self-dimerization body, heterodimer, hairpin, mismatch เป็นต้น สองภาพสุดท้ายในรูปที่ 4 เป็นผลการทดสอบของไพรเมอร์ผลลัพธ์ของไพรเมอร์ด้านหน้านั้นดี ไม่มีโครงสร้างไดเมอร์และกิ๊บที่ชัดเจน ไม่มีฐานเสริมที่ต่อเนื่องกัน และค่าสัมบูรณ์ของพลังงานอิสระน้อยกว่า 4.5 ในขณะที่ไพรเมอร์ด้านหลังแสดงต่อเนื่องกัน ฐาน 6 ฐานประกอบกัน และพลังงานอิสระคือ 8.8;นอกจากนี้ ไดเมอร์ที่ร้ายแรงกว่าจะปรากฏที่ปลาย 3 ′และไดเมอร์ 4 ฐานที่ต่อเนื่องกันจะปรากฏขึ้นแม้ว่าพลังงานอิสระจะไม่สูง แต่ Chl 3 ′dimer สามารถส่งผลกระทบต่อความจำเพาะของการขยายและประสิทธิภาพการขยายอย่างจริงจังนอกจากนี้ จำเป็นต้องตรวจสอบกิ๊บติดผม เฮเทอโรไดเมอร์ และไม่ตรงกัน
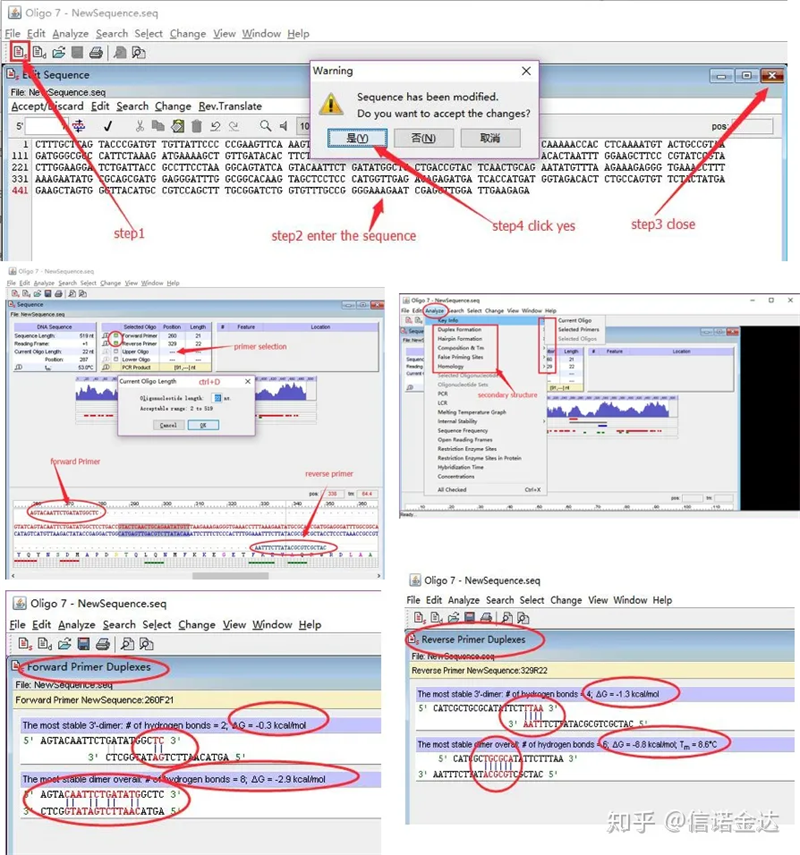
ผลการตรวจจับรูปที่ 3 oligo7
การตรวจจับประสิทธิภาพการขยาย:
ประสิทธิภาพการขยายของปฏิกิริยา PCR ส่งผลกระทบอย่างมากต่อผลลัพธ์ของ PCRนอกจากนี้ ใน qRT-PCR ประสิทธิภาพการขยายสัญญาณยังมีความสำคัญอย่างยิ่งต่อผลลัพธ์เชิงปริมาณนำสาร เครื่องจักร และโปรโตคอลอื่น ๆ ออกจากบัฟเฟอร์ปฏิกิริยาคุณภาพของไพรเมอร์ยังมีอิทธิพลอย่างมากต่อประสิทธิภาพการขยายของ qRT-PCRเพื่อให้มั่นใจถึงความแม่นยำของผลลัพธ์ ทั้งการหาปริมาณฟลูออเรสเซนซ์สัมพัทธ์และการหาปริมาณฟลูออเรสเซนซ์สัมบูรณ์จำเป็นต้องตรวจจับประสิทธิภาพการขยายของไพรเมอร์เป็นที่ทราบกันดีว่าประสิทธิภาพการขยายสัญญาณ qRT-PCR ที่มีประสิทธิภาพอยู่ระหว่าง 85% ถึง 115%มีสองวิธี:
1. วิธีโค้งมาตรฐาน:
ก.ผสม cDNA
ข.การเจือจางแบบไล่ระดับสี
ค.qPCR
ง.สมการถดถอยเชิงเส้นเพื่อคำนวณประสิทธิภาพการขยาย
2. ลินเร็กพีซีอาร์
LinRegPCR เป็นโปรแกรมสำหรับวิเคราะห์ข้อมูล RT-PCR ตามเวลาจริง หรือเรียกอีกอย่างว่าข้อมูล PCR เชิงปริมาณ (qPCR) ตาม SYBR Green หรือเคมีที่คล้ายกันโปรแกรมใช้ข้อมูลที่แก้ไขโดยไม่ใช่เส้นฐาน ดำเนินการแก้ไขเส้นฐานในแต่ละตัวอย่างแยกจากกัน กำหนดหน้าต่างของการเชิงเส้น จากนั้นใช้การวิเคราะห์การถดถอยเชิงเส้นเพื่อให้พอดีกับเส้นตรงผ่านชุดข้อมูล PCRจากความชันของเส้นนี้ จะคำนวณประสิทธิภาพ PCR ของแต่ละตัวอย่างประสิทธิภาพ PCR เฉลี่ยต่อแอมพลิคอนและค่า Ct ต่อตัวอย่างใช้ในการคำนวณความเข้มข้นเริ่มต้นต่อตัวอย่าง ซึ่งแสดงเป็นหน่วยการเรืองแสงตามอำเภอใจอินพุตและเอาต์พุตข้อมูลผ่านสเปรดชีต Excelเฉพาะตัวอย่าง
จำเป็นต้องผสมไม่มีการไล่ระดับสี
จำเป็นต้องมีขั้นตอน:(ยกตัวอย่าง Bole CFX96 ไม่ใช่เครื่องที่มี ABI ชัดเจน)
การทดลอง:เป็นการทดลอง qPCR มาตรฐาน
เอาต์พุตข้อมูล qPCR:LinRegPCR สามารถจดจำไฟล์เอาต์พุตได้สองรูปแบบ: RDML หรือผลลัพธ์การขยายปริมาณในความเป็นจริง มันคือค่าการตรวจจับตามเวลาจริงของหมายเลขรอบและสัญญาณการเรืองแสงโดยเครื่องจักร และการขยายได้มาจากการวิเคราะห์ค่าการเปลี่ยนแปลงการเรืองแสงของประสิทธิภาพของเซกเมนต์เชิงเส้น
การเลือกข้อมูล: ตามทฤษฎีแล้ว ค่า RDML ควรจะใช้งานได้ประมาณว่าปัญหาของคอมพิวเตอร์ของฉันคือซอฟต์แวร์ไม่รู้จัก RDML ดังนั้นฉันจึงมีค่าเอาต์พุต excel เป็นข้อมูลต้นฉบับขอแนะนำให้ทำการคัดกรองข้อมูลอย่างคร่าวๆ ก่อน เช่น ความล้มเหลวในการเพิ่มตัวอย่าง เป็นต้น จุดสามารถลบได้ในข้อมูลเอาต์พุต (แน่นอน คุณไม่สามารถลบได้ LinRegPCR จะเพิกเฉยต่อจุดเหล่านี้ในขั้นต่อมา)
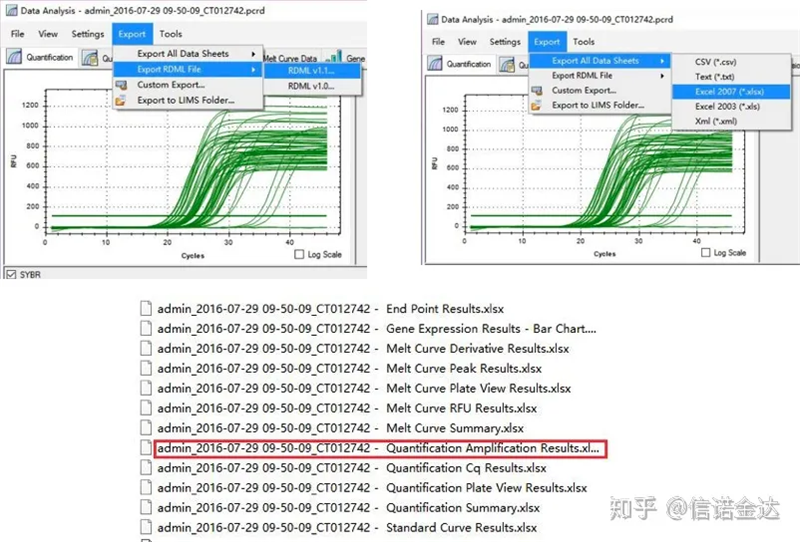
รูปที่ 5 การส่งออกข้อมูล qPCR
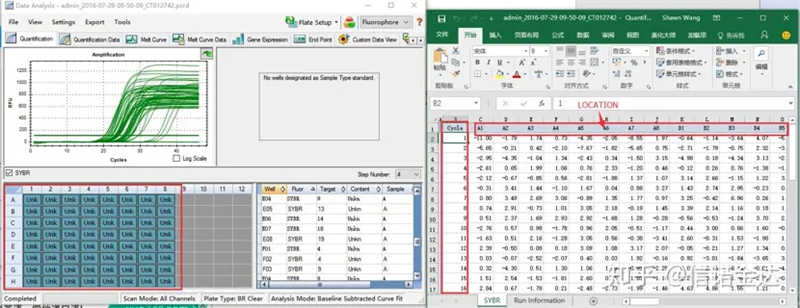
รูปที่ 6 การเลือกตัวอย่างผู้สมัคร
ป้อนข้อมูล:เปิดคุณสมบัติการขยายไฟล์ xls, → เปิด LinRegPCR → ไฟล์ → อ่านจาก excel → เลือกพารามิเตอร์ดังรูปที่ 7 → ตกลง → คลิก กำหนดเส้นฐาน
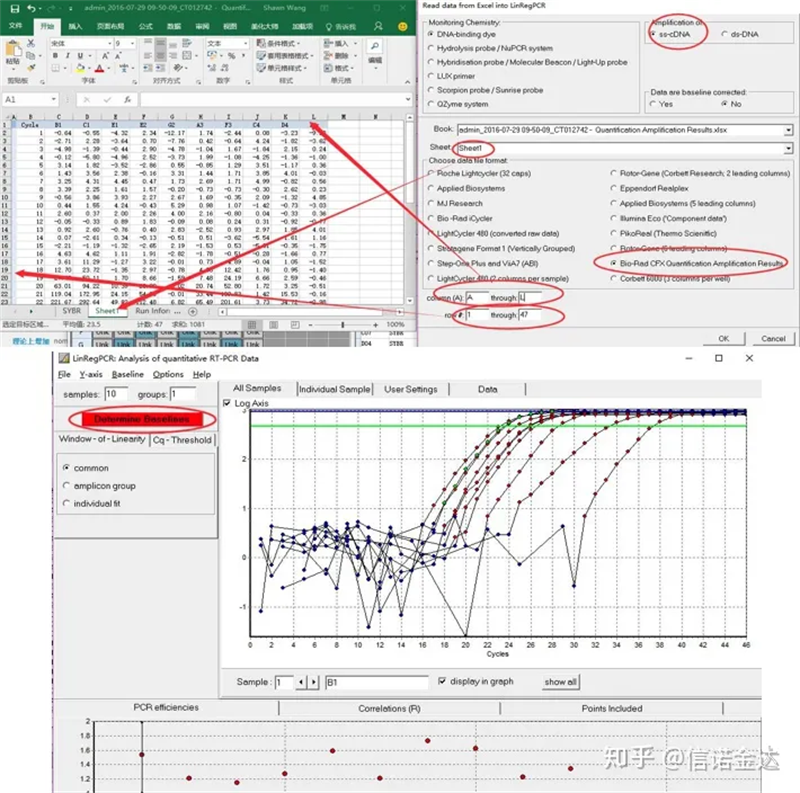
รูปที่ 7 ขั้นตอนของการป้อนข้อมูล linRegPCR
ผลลัพธ์:หากไม่มีการทำซ้ำก็ไม่จำเป็นต้องจัดกลุ่มหากมีการทำซ้ำ การจัดกลุ่มสามารถแก้ไขได้ในการจัดกลุ่มตัวอย่าง และป้อนชื่อยีนในตัวระบุ จากนั้นยีนเดียวกันจะถูกจัดกลุ่มโดยอัตโนมัติสุดท้าย คลิกที่ไฟล์ ส่งออก excel และดูผลลัพธ์ประสิทธิภาพการขยายและผลลัพธ์ R2 ของแต่ละหลุมจะแสดงขึ้นประการที่สอง หากคุณแบ่งออกเป็นกลุ่ม ประสิทธิภาพการขยายเฉลี่ยที่แก้ไขแล้วจะแสดงขึ้นตรวจสอบให้แน่ใจว่าประสิทธิภาพการขยายของไพรเมอร์แต่ละตัวอยู่ระหว่าง 85% ถึง 115%หากใหญ่หรือเล็กเกินไป แสดงว่าประสิทธิภาพการขยายของไพรเมอร์ไม่ดี
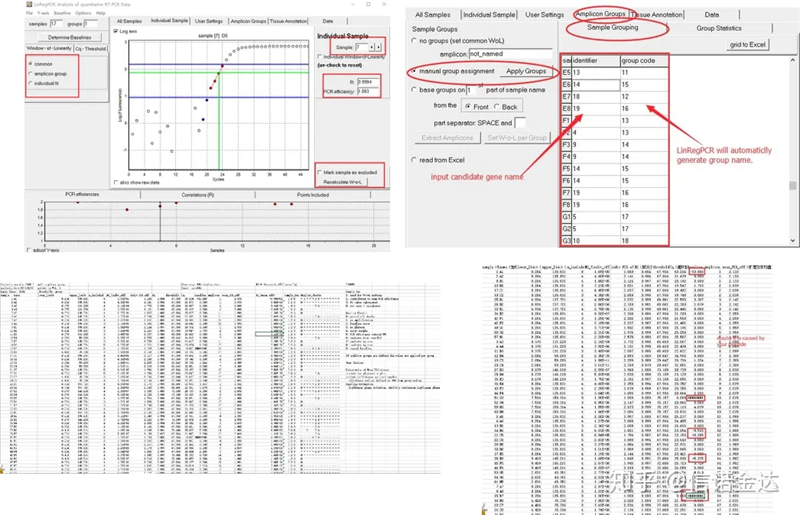
รูปที่ 8 ผลลัพธ์และข้อมูลออก
กระบวนการทดลอง:
ข้อกำหนดด้านคุณภาพ RNA:
ความบริสุทธิ์:1.72.0 บ่งชี้ว่าอาจมีไอโซไทโอไซยาเนตตกค้างกรดนิวคลีอิกที่สะอาด A260/A230 ควรอยู่ที่ประมาณ 2 หากมีการดูดกลืนที่รุนแรงที่ 230 นาโนเมตร แสดงว่ามีสารประกอบอินทรีย์ เช่น ฟีเนตไอออนนอกจากนี้ยังสามารถตรวจจับได้ด้วย agarose gel electrophoresis 1.5%ชี้เครื่องหมาย เนื่องจาก ssRNA ไม่มีการเสียสภาพธรรมชาติและลอการิทึมน้ำหนักโมเลกุลไม่มีความสัมพันธ์เชิงเส้น และไม่สามารถแสดงน้ำหนักโมเลกุลได้อย่างถูกต้องความเข้มข้น: ในทางทฤษฎีไม่น้อยกว่า 100ng/ul ถ้าความเข้มข้นต่ำเกินไป ความบริสุทธิ์โดยทั่วไปจะต่ำไม่สูง
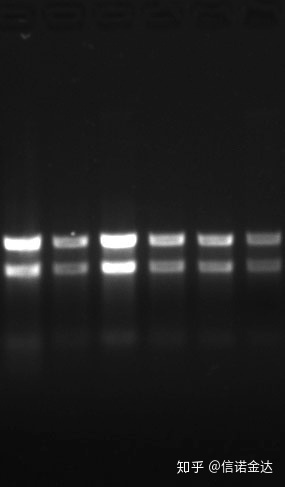
ฟิก9 อาร์เอ็นเอ เจล
นอกจากนี้ หากตัวอย่างมีค่าและความเข้มข้นของ RNA สูง ขอแนะนำให้แบ่งส่วนหลังการสกัด และเจือจาง RNA ให้ได้ความเข้มข้นสุดท้ายที่ 100-300ng/ul สำหรับการถอดความแบบย้อนกลับในกระบวนการถอดความแบบย้อนกลับเมื่อ mRNA ถูกถอดความ ไพรเมอร์ oligo (dt) ที่สามารถจับกับหางของ polyA โดยเฉพาะจะใช้สำหรับการถอดรหัสแบบย้อนกลับ ในขณะที่ lncRNA และ circRNA ใช้ไพรเมอร์ hexamer แบบสุ่ม (สุ่ม 6 mer) สำหรับการถอดรหัสแบบย้อนกลับของ RNA ทั้งหมด สำหรับ miRNA จะใช้ไพรเมอร์แบบวนรอบคอเฉพาะของ miRNA สำหรับการถอดรหัสแบบย้อนกลับหลายบริษัทได้เปิดตัวชุดหางปลาแบบพิเศษแล้วสำหรับวิธีสเต็มลูป วิธีหางแร่นั้นสะดวกกว่า ปริมาณงานสูง และประหยัดรีเอเจนต์ แต่ผลของการแยกแยะ miRNA ของตระกูลเดียวกันไม่น่าจะดีเท่าวิธีสเต็มลูปชุดการถอดความแบบย้อนกลับแต่ละชุดมีข้อกำหนดสำหรับความเข้มข้นของไพรเมอร์เฉพาะยีน (สเต็มลูป)การอ้างอิงภายในที่ใช้สำหรับ miRNA คือ U6ในกระบวนการกลับด้านของสเต็มลูป ควรกลับท่อของ U6 แยกกัน และควรเติมไพรเมอร์ด้านหน้าและด้านหลังของ U6 โดยตรงทั้ง circRNA และ lncRNA สามารถใช้ HKG เป็นข้อมูลอ้างอิงภายในได้ในการตรวจจับ cDNA,
ถ้าไม่มีปัญหากับ RNA ก็ควรใช้ cDNA เช่นกันอย่างไรก็ตาม หากติดตามความสมบูรณ์แบบของการทดลอง วิธีที่ดีที่สุดคือใช้ยีนอ้างอิงภายใน (ยีนอ้างอิง, RG) ที่สามารถแยกความแตกต่างของ gDNA จากซีดีโดยทั่วไป RG เป็นยีนดูแลทำความสะอาด, HKG) ดังรูปที่ 10;ในเวลานั้น ฉันกำลังสร้างโปรตีนกักเก็บถั่วเหลือง และใช้แอคติน 7 ที่มีอินตรอนเป็นข้อมูลอ้างอิงภายในขนาดของส่วนขยายของไพรเมอร์นี้ใน gDNA คือ 452bp และถ้าใช้ cDNA เป็นแม่แบบ ก็จะเท่ากับ 142bpจากนั้นผลการทดสอบพบว่าส่วนหนึ่งของ cDNA ถูกปนเปื้อนโดย gDNA และยังพิสูจน์ได้ว่าไม่มีปัญหากับผลการถอดความแบบย้อนกลับ และสามารถใช้เป็นแม่แบบสำหรับ PCRมันไม่มีประโยชน์ที่จะเรียกใช้ agarose gel electrophoresis โดยตรงกับ cDNA และมันเป็นแถบกระจายซึ่งไม่น่าเชื่อ
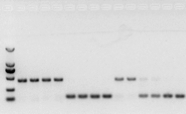
รูปที่ 10 การตรวจจับ cDNA
การกำหนดเงื่อนไข qPCRโดยทั่วไปไม่มีปัญหาตามโปรโตคอลของชุด ส่วนใหญ่อยู่ในขั้นตอนของค่า tmหากไพรเมอร์บางตัวได้รับการออกแบบมาไม่ดีในระหว่างการออกแบบไพรเมอร์ ส่งผลให้ค่า tm และ 60°C ตามทฤษฎีแตกต่างกันมาก แนะนำให้ใช้ cDNA หลังจากผสมตัวอย่างแล้ว ให้รัน PCR แบบไล่ระดับสีด้วยไพรเมอร์ และพยายามหลีกเลี่ยงการตั้งอุณหภูมิโดยไม่มีแถบเป็นค่า TM
การวิเคราะห์ข้อมูล
วิธีการประมวลผล PCR เชิงปริมาณเรืองแสงสัมพัทธ์แบบธรรมดานั้นเป็นไปตาม 2-ΔΔCT.แม่แบบการประมวลผลข้อมูล
สินค้าที่เกี่ยวข้อง:
PCR แบบเรียลไทม์ง่ายTM – แท็กแมน
PCR แบบเรียลไทม์ง่ายTM –SYBR กรีน
RT Easy I (มาสเตอร์พรีมิกซ์สำหรับการสังเคราะห์ cDNA สายแรก)
RT Easy II (มาสเตอร์พรีมิกซ์สำหรับการสังเคราะห์ cDNA สายแรกสำหรับ qPCR)
เวลาโพสต์: Mar-14-2023



