



PCR (ปฏิกิริยาลูกโซ่โพลีเมอเรส) เป็นหนึ่งในเทคโนโลยีการขยาย DNA ในหลอดทดลองที่มีประวัติยาวนานกว่า 30 ปี
เทคโนโลยี PCR ได้รับการบุกเบิกโดย Kary Mullis แห่ง Cetus ประเทศสหรัฐอเมริกาในปี 1983 Mullis ยื่นขอสิทธิบัตร PCR ในปี 1985 และตีพิมพ์บทความวิชาการ PCR ฉบับแรกเกี่ยวกับ Science ในปีเดียวกันมัลลิสได้รับรางวัลโนเบลสาขาเคมีในปี 1993 จากผลงานของเขา
หลักการพื้นฐานของ PCR
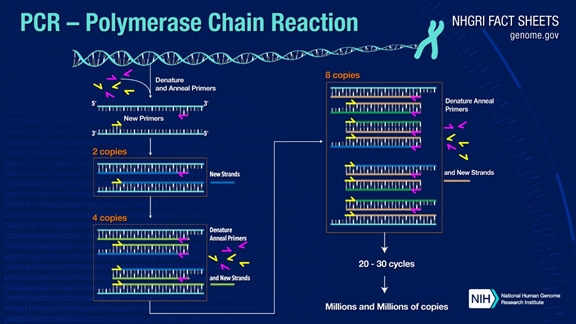
PCR สามารถขยายชิ้นส่วน DNA เป้าหมายได้มากกว่าหนึ่งล้านเท่าหลักการนี้อยู่ภายใต้การเร่งปฏิกิริยาของ DNA polymerase โดยใช้ DNA สายหลักเป็นแม่แบบและไพรเมอร์เฉพาะเป็นจุดเริ่มต้นสำหรับการขยายมีการทำซ้ำในหลอดทดลองผ่านขั้นตอนต่าง ๆ เช่น การสูญเสียสภาพธรรมชาติ การหลอม และการขยายกระบวนการของ DNA สายลูกสาวเสริมกับ DNA แม่แบบสายแม่

กระบวนการ PCR มาตรฐานแบ่งออกเป็นสามขั้นตอน:
1.Denaturation: ใช้อุณหภูมิสูงในการแยกสายคู่ของ DNAพันธะไฮโดรเจนระหว่างสายคู่ของ DNA ถูกทำลายที่อุณหภูมิสูง (93-98 ℃)
2.การหลอม: หลังจากแยก DNA สายคู่แล้ว ให้ลดอุณหภูมิลงเพื่อให้ไพรเมอร์จับกับ DNA สายเดี่ยวได้
3. ส่วนขยาย: DNA polymerase เริ่มสังเคราะห์สายเสริมตามสาย DNA จากไพรเมอร์ที่จับกันเมื่ออุณหภูมิลดลงเมื่อการขยายเสร็จสิ้น วัฏจักรจะเสร็จสมบูรณ์ และจำนวนชิ้นส่วนของ DNA จะเพิ่มเป็นสองเท่า
การดำเนินการทั้งสามขั้นตอนนี้ซ้ำกัน 25-35 ครั้ง จำนวนชิ้นส่วน DNA จะเพิ่มขึ้นอย่างทวีคูณ
ความฉลาดของ PCR คือไพรเมอร์ที่แตกต่างกันสามารถออกแบบสำหรับยีนเป้าหมายที่แตกต่างกัน เพื่อให้ชิ้นส่วนของยีนเป้าหมายสามารถขยายได้ในระยะเวลาอันสั้น
จนถึงขณะนี้ PCR สามารถแบ่งออกได้เป็น 3 ประเภท ได้แก่ PCR ธรรมดา PCR เชิงปริมาณเรืองแสง และ PCR ดิจิทัล
PCR สามัญรุ่นแรก
ใช้เครื่องมือขยาย PCR ทั่วไปเพื่อขยายยีนเป้าหมาย จากนั้นใช้ agarose gel electrophoresis เพื่อตรวจจับผลิตภัณฑ์ ทำได้เฉพาะการวิเคราะห์เชิงคุณภาพเท่านั้น
ข้อเสียเปรียบหลักของ PCR รุ่นแรก:
1. มีแนวโน้มที่จะขยายแบบไม่เฉพาะเจาะจงและผลบวกปลอม
2. การตรวจจับใช้เวลานานและการดำเนินการยุ่งยาก
3. ทำได้เฉพาะการทดสอบเชิงคุณภาพเท่านั้น
PCR แบบเรียลไทม์รุ่นที่สอง
Real-Time PCR หรือที่เรียกว่า qPCR ใช้โพรบเรืองแสงที่สามารถระบุความคืบหน้าของระบบปฏิกิริยา และตรวจสอบการสะสมของผลิตภัณฑ์ที่ขยายผ่านการสะสมของสัญญาณเรืองแสง และตัดสินผลลัพธ์ผ่านเส้นโค้งเรืองแสงสามารถหาปริมาณได้โดยใช้ค่า Cq และเส้นโค้งมาตรฐาน
เนื่องจากเทคโนโลยี qPCR ดำเนินการในระบบปิด ความน่าจะเป็นของการปนเปื้อนจึงลดลง และสามารถตรวจสอบสัญญาณเรืองแสงสำหรับการตรวจจับเชิงปริมาณได้ ดังนั้นจึงใช้กันอย่างแพร่หลายในการปฏิบัติทางคลินิกและกลายเป็นเทคโนโลยีที่โดดเด่นใน PCR
สารเรืองแสงที่ใช้ใน PCR เชิงปริมาณเรืองแสงตามเวลาจริงสามารถแบ่งออกเป็น: โพรบเรืองแสง TaqMan, สัญญาณบอกระดับโมเลกุล และสีย้อมเรืองแสง
1) หัววัดเรืองแสง TaqMan:
ในระหว่างการขยาย PCR จะมีการเพิ่มโพรบเรืองแสงเฉพาะในขณะที่เพิ่มไพรเมอร์หนึ่งคู่หัววัดเป็นโอลิโกนิวคลีโอไทด์ และปลายทั้งสองด้านมีกลุ่มเรืองแสงรีพอร์เตอร์และกลุ่มเรืองแสงดับ
เมื่อโพรบไม่เสียหาย สัญญาณเรืองแสงที่ปล่อยออกมาจากกลุ่มนักข่าวจะถูกดูดซับโดยกลุ่มดับในระหว่างการขยาย PCR กิจกรรม exonuclease 5′-3′ ของเอนไซม์ Taq จะแยกและย่อยสลายโพรบ ทำให้กลุ่มเรืองแสงของนักข่าวและสารดับ กลุ่มเรืองแสงถูกแยกออกจากกัน เพื่อให้ระบบตรวจสอบการเรืองแสงสามารถรับสัญญาณเรืองแสงได้ นั่นคือทุกครั้งที่มีการขยายสาย DNA โมเลกุลเรืองแสงจะก่อตัวขึ้น และการสะสมของสัญญาณเรืองแสงจะประสานกับการก่อตัวของผลิตภัณฑ์ PCR อย่างสมบูรณ์
2) สีย้อมเรืองแสง SYBR:
ในระบบปฏิกิริยา PCR มีการเติมสีย้อมเรืองแสง SYBR ที่มากเกินไปหลังจากที่สีย้อมเรืองแสง SYBR ไม่ถูกรวมเข้ากับเส้นใยคู่ของ DNA อย่างเฉพาะเจาะจง มันจะส่งสัญญาณเรืองแสงโมเลกุลสีย้อม SYBR ที่ไม่ได้รวมอยู่ในสายโซ่จะไม่ปล่อยสัญญาณเรืองแสงใด ๆ ดังนั้นจึงมั่นใจได้ว่าสัญญาณเรืองแสง การเพิ่มขึ้นของผลิตภัณฑ์ PCR จะซิงโครไนซ์กับการเพิ่มขึ้นของผลิตภัณฑ์ PCR อย่างสมบูรณ์SYBR จับกับ DNA ที่มีเกลียวสองเส้นเท่านั้น ดังนั้นจึงสามารถใช้เส้นกราฟการหลอมเหลวเพื่อตรวจสอบว่าปฏิกิริยา PCR นั้นจำเพาะหรือไม่

3) สัญญาณเตือนระดับโมเลกุล:
เป็นโพรบโอลิโกนิวคลีโอไทด์ที่มีฉลากติดฉลากสองครั้งที่ก้านซึ่งสร้างโครงสร้างกิ๊บประมาณ 8 ฐานที่ปลาย 5 และ 3ลำดับกรดนิวคลีอิกที่ปลายทั้งสองจับคู่กัน ทำให้กลุ่มเรืองแสงและกลุ่มดับแน่นปิด จะไม่มีการผลิตสารเรืองแสง

หลังจากสร้างผลิตภัณฑ์ PCR แล้ว ในระหว่างกระบวนการหลอม ส่วนตรงกลางของสัญญาณโมเลกุลจะถูกจับคู่กับลำดับดีเอ็นเอเฉพาะ และยีนเรืองแสงจะถูกแยกออกจากยีนดับเพื่อผลิตสารเรืองแสง

ข้อเสียเปรียบหลักของ PCR รุ่นที่สอง:
ความไวยังขาดอยู่และการตรวจจับตัวอย่างสำเนาต่ำนั้นไม่แม่นยำ
มีอิทธิพลของค่าพื้นหลัง และผลลัพธ์จะไวต่อสัญญาณรบกวน
เมื่อมีสารยับยั้ง PCR ในระบบปฏิกิริยา ผลการตรวจจับจะไวต่อการรบกวน
PCR ดิจิทัลรุ่นที่สาม
Digital PCR (DigitalPCR, dPCR, Dig-PCR) คำนวณหมายเลขสำเนาของลำดับเป้าหมายผ่านการตรวจจับจุดสิ้นสุด และสามารถทำการตรวจจับเชิงปริมาณที่แม่นยำโดยไม่ต้องใช้การควบคุมภายในและเส้นโค้งมาตรฐาน
Digital PCR ใช้การตรวจจับจุดสิ้นสุดและไม่ขึ้นอยู่กับค่า Ct (เกณฑ์รอบ) ดังนั้นปฏิกิริยา PCR แบบดิจิทัลจึงได้รับผลกระทบน้อยลงจากประสิทธิภาพการขยายสัญญาณ และความทนทานต่อสารยับยั้งปฏิกิริยา PCR จึงดีขึ้น โดยมีความแม่นยำและความสามารถในการทำซ้ำสูง
เนื่องจากลักษณะเฉพาะของความไวสูงและความแม่นยำสูง จึงไม่ถูกแทรกแซงโดยสารยับยั้งปฏิกิริยา PCR อย่างง่ายดาย และสามารถบรรลุปริมาณสัมบูรณ์ที่แท้จริงโดยไม่ต้องใช้ผลิตภัณฑ์มาตรฐาน ซึ่งกลายเป็นฮอตสปอตการวิจัยและการประยุกต์ใช้
ตามรูปแบบต่างๆ ของหน่วยปฏิกิริยา สามารถแบ่งออกได้เป็นสามประเภทหลัก ได้แก่ ระบบไมโครฟลูอิดิก ระบบชิป และระบบหยด
1) PCR ดิจิตอลไมโครฟลูอิดิก, mdPCR:
ด้วยเทคโนโลยีไมโครฟลูอิดิก แม่แบบ DNA จะถูกแยกออกจากกันเทคโนโลยีไมโครฟลูอิดิกสามารถรับรู้ถึงการอัปเกรดนาโนของตัวอย่างหรือการสร้างหยดขนาดเล็กลง แต่หยดนั้นต้องการวิธีการดูดซับแบบพิเศษ แล้วจึงรวมเข้ากับระบบปฏิกิริยา PCRmdPCR ค่อยๆ ถูกนำมาใช้โดยวิธีอื่นแทนที่
2) PCR ดิจิทัลแบบหยดตาม ddPCR:
ใช้เทคโนโลยีการสร้างหยดน้ำในน้ำมันเพื่อประมวลผลตัวอย่างเป็นหยด และแบ่งระบบปฏิกิริยาที่มีโมเลกุลของกรดนิวคลีอิกออกเป็นหยดขนาดนาโนหลายพันหยด ซึ่งแต่ละหยดไม่มีโมเลกุลเป้าหมายของกรดนิวคลีอิกที่จะตรวจพบ หรือมีโมเลกุลเป้าหมายของกรดนิวคลีอิกตั้งแต่หนึ่งถึงหลายตัวที่จะทดสอบ
3) PCR ดิจิทัลที่ใช้ชิป, cdPCR:
ใช้เทคโนโลยีทางเดินของเหลวในตัวเพื่อแกะสลักท่อขนาดเล็กและโพรงขนาดเล็กจำนวนมากบนแผ่นเวเฟอร์ซิลิคอนหรือแก้วควอทซ์ และควบคุมการไหลของสารละลายผ่านวาล์วควบคุมต่างๆ และแบ่งของเหลวตัวอย่างเป็นนาโนเมตรที่มีขนาดเท่ากันในหลุมปฏิกิริยาสำหรับปฏิกิริยา PCR แบบดิจิทัลเพื่อให้ได้ปริมาณที่แน่นอน
ข้อเสียเปรียบหลักของ PCR รุ่นที่สาม:
อุปกรณ์และน้ำยามีราคาแพง
ข้อกำหนดด้านคุณภาพของเทมเพลตอยู่ในระดับสูงหากปริมาณเท็มเพลตเกินปริมาณของไมโครซิสเต็ม จะไม่สามารถวัดปริมาณได้ และหากมีปริมาณน้อยเกินไป ความแม่นยำในการหาปริมาณจะลดลง
นอกจากนี้ยังสามารถสร้างผลบวกลวงได้เมื่อมีการขยายที่ไม่เฉพาะเจาะจง
เวลาโพสต์: กรกฎาคม-30-2021



